
Improving the catalytic activity of Au/Pd core–shell nanoparticles with a tailored Pd structure for formic acid oxidation reaction
Abstract
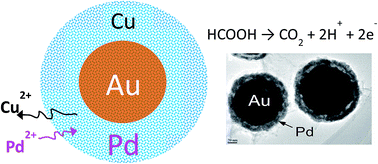
Unique morphology-tunable Au/Pd core–shell nanoparticles were synthesized by galvanic replacement of preformed Cu on hollow Au cores using different PdCl2 concentrations. The nanoparticles were characterized by X-ray diffraction (XRD), transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS), and electrochemical analysis. The results showed that the structure of the nanocrystalline Pd on the hollow Au core surface was strongly dependent on the PdCl2 concentration. It was found that Pd2+ ions transport and react in the porous Cu layer, helping to create a continuous but porous structure which enlarges the Pd surface area and increases the electrochemical activity. In addition, the Au/Pd core–shell nanoparticles displayed superior electrochemical performance and stability in formic acid oxidation than commercial Pd black, especially for the ones synthesized using 2.5 mM PdCl2. The enhanced electrocatalytic performance may be attributed to the optimum electronic coupling effect caused by the interaction between the specific Pd structure and the hollow Au core.
 Please wait while we load your content...
Please wait while we load your content...

