
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn unusual one-donor-two-acceptor interaction in a pair of covalently bridged naphthalenediimide dimers†
Soumik
Sao
,
Bibek R.
Samanta
and
Debangshu
Chaudhuri
*
Department of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Kolkata, Mohanpur 741246, India. E-mail: dchaudhuri@iiserkol.ac.in
First published on 31st March 2016
Abstract
An unusual donor–acceptor (D–A) state is reported in a pair of regioisomeric xylylene-bridged naphthalenediimide (NDI) dimers. The D–A state is characterized by a weak, red-shifted absorbance and a photoluminescence (PL) with a very high quantum efficiency. PL and PL excitation (PLE) spectroscopy reveals the partial charge-transfer character of the D–A state. Disruption of this D–A interaction in the presence of a competing donor, reveals the unique one-donor-two-acceptors stoichiometry. Finally, the effect of intramolecular pi-stacking on the D–A interaction is discussed.
Multichromophoric aggregates garner a lot of attention as promising candidates for electronic and photovoltaic applications.1 In an aggregate, strong excitonic interactions between multiple chromophores give rise to a variety of fascinating phenomena, such as exciton migration2 and charge transfer.3 Covalently-bridged bi- or tri-chromophoric compounds are often used as model systems to gain a precise understanding of interchromophoric interactions and their influence on the electronic properties of molecular aggregates.4 Since in these model systems, the focus is primarily on interchromophoric interactions, the bridging unit only serves as a structural motif that predisposes the chromophores to a specific orientation. An alternative, but less explored design strategy is the one, where the bridge unit itself participates in a specific electronic interaction with the chromophore. By inducing aggregation between the chromophores, one can then study how the “bridge-chromophore” interaction evolves into a “bridge-aggregate” type interaction. This can be particularly relevant in the context of D–A type aggregates, where the D–A interface is constructed by coating D (or A) molecules onto the surface of a preassembled A (or D) nanostructures.5 In such systems, the key interaction is between the molecules of one kind (D or A) and the aggregate of the other (A or D).
NDIs have emerged as one of most promising organic n-type semiconductors.6 Its highly electron-deficient core, on one hand facilitates molecular aggregation through pi-stacking, and on the other, serves as a versatile acceptor unit in various D–A geometries.7 We report a pair of regioisomeric NDI dimer systems, 1 and 2, where the two electron-deficient NDI units are linked through a moderately electron-rich xylylene bridge, creating the possibility of a D–A type interaction. In addition to these acceptor–donor–acceptor (A–D–A) triads, we also have included 3, a D–A dyad in our study (see Scheme 1). There are recent reports on D–A type interactions between NDI and aromatic solvent molecules that lead to a photoluminescent state.8 Photoluminescence (PL) is also reported from the interaction of NDI and aromatic guest molecules in a confined metal–organic framework.9 Thus, the prospect of having an emissive D–A state makes it convenient to monitor NDI–xylylene interaction in our compounds. Further, in a suitable solvent environment, molecule 1 can fold into a conformation that allows the two NDI units to stack cofacially, forming an intramolecular H-type aggregate. This provides the opportunity to monitor the fate of D–A interaction between xylylene donor and the NDI dimeric aggregate. 1 and 2 were synthesized in a two-step process, by condensation of a pre-synthesized naphthalene monoimide 4 with the corresponding diamine (see Scheme 1 and ESI† for details).10
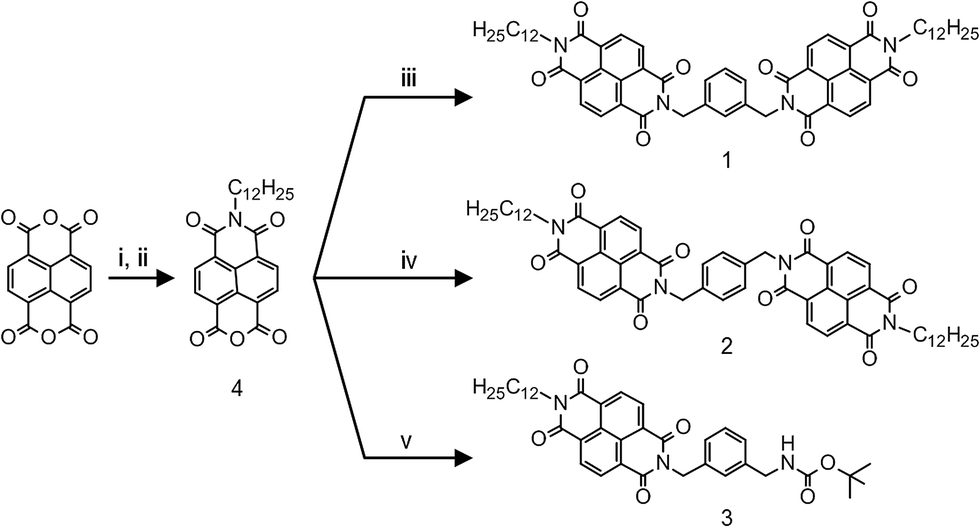 | ||
Scheme 1 Syntheses of 1, 2 and 3. (i) C12H25NH2, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 iPrOH/H2O, 70 °C, Ar; (ii) glacial AcOH, reflux; (iii) m-xylenediamine, dry DMF, 120 °C, Ar; (iv) p-xylenediamine, dry DMF, 120 °C, Ar; (v) mono-N-Boc-m-xylenediamine, dry DMF, 120 °C, Ar. :1 iPrOH/H2O, 70 °C, Ar; (ii) glacial AcOH, reflux; (iii) m-xylenediamine, dry DMF, 120 °C, Ar; (iv) p-xylenediamine, dry DMF, 120 °C, Ar; (v) mono-N-Boc-m-xylenediamine, dry DMF, 120 °C, Ar. | ||
Optical absorption spectra of 1, 2 and 3 are presented in Fig. 1a. The spectra were recorded in dilute dichloromethane solutions (∼10 μM) in order to avoid any intermolecular effects.‡ The dominant absorption band in all three cases is identical, characterized by vibronic progressions at 380, 360 and 340 nm, typical of an isolated NDI chromophore.10 Evidently, in a good solvent such as CH2Cl2, there is no pi-stacking between the NDI units. In addition to the absorption band of the non-interacting NDI chromophore, a weak and strongly red-shifted feature at 510 and 475 nm appears in the spectrum of the m-xylylene dimer, 1. A closer look at 400–550 nm wavelength range (see inset) reveals that the weak absorption feature is also present, slightly broadened in the p-xylylene dimer, 2, but is completely absent in 3. Identical absorption spectra were measured in THF,† which proves that the red-shifted absorption feature is not a consequence of any halogen–pi interaction involving CH2Cl2. We attribute this absorption feature to a donor–acceptor (D–A) interaction between the aromatic ring of the xylylene bridge and the NDI units. Lack of a similar D–A interaction in the dyad, 3, and its characteristic but an unprecedented red-shifted (∼100 nm) absorbance are clear indications that the nature of xylylene–NDI interaction in 1 and 2 is markedly different from earlier instances of D–A interactions involving NDI and aromatic compounds.8,9 Subsequent PL and PLE spectroscopy results illustrate how this weakly absorbing D–A state exclusively dominates the photophysics of the two dimers.
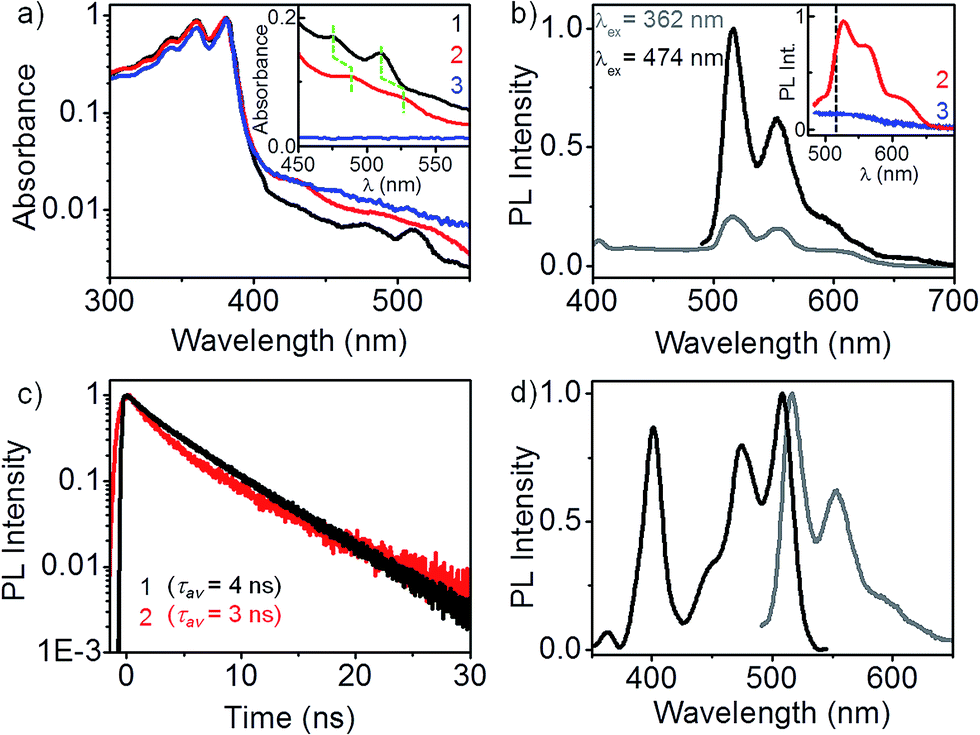 | ||
| Fig. 1 Donor–acceptor state of the dimers. (a) Absorption spectra of 1, 2 and 3 in CH2Cl2 (∼10 μM). Characteristic NDI absorption appears below 400 nm. Additionally in 1 and 2, a weak absorption feature appears between 475–525 nm that corresponds to a D–A interaction. Inset: spectra recorded at ∼0.5 mM‡ clearly show the absence of D–A species in 3. (b) PL is exclusively from the D–A state, even when the excitation is carried out in the dominant higher energy absorption band of NDI chromophore. Inset: PL spectra of 2 (λex = 474 nm) is similar to that of 1 (peak position marked with a black dashed line), but 11 nm red-shifted. Monomeric 3 shows no PL at 474 nm excitation, (c) long-lived (τav = 3–4 ns) PL from 1 and 2 is consistent with the D–A nature of the emissive state, (d) PLE spectrum (λPL = 555 nm) of 1 bears a perfect mirror-image symmetry to the PL spectrum, with a near-negligible stokes shift. | ||
PL spectra of 1 (Fig. 1b) in dilute CH2Cl2 solutions presents several interesting aspects. When excited in the dominant absorption band of the non-interacting NDI chromophore (at 362 nm), no PL is observed between 400 and 430 nm, a region where NDI emission is likely to appear. Instead, a vibronically-resolved PL spectrum appears in 510–570 nm region. Surprisingly, the intensity of 510 nm PL increases many folds when excited at 474 nm, where the molecular absorption cross-section is more than two orders of magnitude smaller. Clearly, the emissive state of dimer 1 is associated with the weakly absorbing D–A species, discussed earlier. The PL quantum efficiency was estimated to be ∼78%.† We further note that the PL spectrum displays a near-negligible stokes shift, a fact more clearly illustrated by PLE spectroscopy. Distinct vibronic features in the PL spectrum at 510, 565 and 600 nm points to a well-defined emissive state that is strongly Franck–Condon coupled to the molecular vibrational modes. This is unlike a typical charge-transfer state which is characterized by a broad, featureless PL spectrum.11 A modest hypsochromic shift† of the PL spectrum in non-polar CCl4 reveals that the emissive state has only a partial charge-transfer character.12 The inset presents the PL spectrum of 2 and 3 at 474 nm excitation. The p-xylylene dimer 2 exhibits a similar but a further red-shifted (∼11 nm) PL spectrum, suggesting that the nature of D–A interaction in both the regioisomers are similar. More importantly, 3 shows no PL emission in 500–600 nm range, confirming the absence of a similar D–A interaction. This leads us to speculate that the D–A interaction in 1 and 2 involves one donor and a pair of acceptors, an interaction not feasible in 3.
PL of 1 and 2 have a nearly identical decay profile with a lifetime of 3–4 ns (Fig. 1c), in sharp contrast to a much shorter PL lifetime known for an isolated NDI chromophore (∼20 ps).13 A longer PL lifetime is consistent with the partial charge-transfer character of the emissive state. The fact that the emission is almost entirely from the D–A species allows us to exclusively investigate it using PLE spectroscopy, without any interference from the dominant absorption of the NDI chromophore. There are two distinct excitation features in the PLE spectrum of 1 (see Fig. 1d): a low energy feature with vibronic progressions at 508 and 474 nm, and a higher energy feature at 400 nm. The lower energy PLE matches perfectly well with the weak absorption band (inset Fig. 1a). Mirror-image symmetry between the lower energy PLE and the PL spectrum, along with an exceedingly small stokes shift of 343 cm−1 point at the structural integrity of the emissive state. The vibronic structure in PL, lower energy PLE, and the dominant band in the absorption spectra bears a common trait: the spacing between successive vibronic bands matches well with the ∼1650 cm−1 asymmetric C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond stretching mode† in NDI.
O bond stretching mode† in NDI.
The nature of D–A interaction in 1 is nicely illustrated by F− binding studies in CH2Cl2.† Electron-deficient NDI displays a strong tendency towards F− binding by means of anion-pi interaction.14 This presents a scenario where two donors, F− and the aromatic ring of the xylylene bridge, compete for the same acceptor, NDI. Even at a very low F− concentration (9 mol%), 510 nm PL emission from the D–A state of 1 is quenched by ∼90% (see Fig. 2b). With subsequent increase in F− concentration, the 510 nm PL is completely quenched, along with the appearance of a new emission at 430 nm. PLE spectrum (inset) confirms that the 430 nm PL is from an isolated NDI chromophore. Since F− is a better donor than the xylylene ring, NDI preferentially forms an anion-pi complex with F− at the expense of D–A interaction with xylylene bridge (see Fig. 2a). Thus, in the limit of very low F− concentration, for every 1:1 NDI–F− complex that forms, the second NDI unit of 1 becomes free and contributes to a PL at 430 nm. It is instructive to note that the free NDI that is generated is however incapable of forming a D–A complex with the xylylene unit. This once again corroborates our earlier hypothesis that the D–A interaction in the dimers involves one xylylene donor and a pair of NDI acceptor units. Fig. 2c presents the corresponding changes in the absorption spectrum of 1. At 9 mol% of F−, the weakly absorbing D–A species is completely replaced by a new absorption feature at 470 nm that corresponds to the formation of 1:1 NDI–F− complex.14
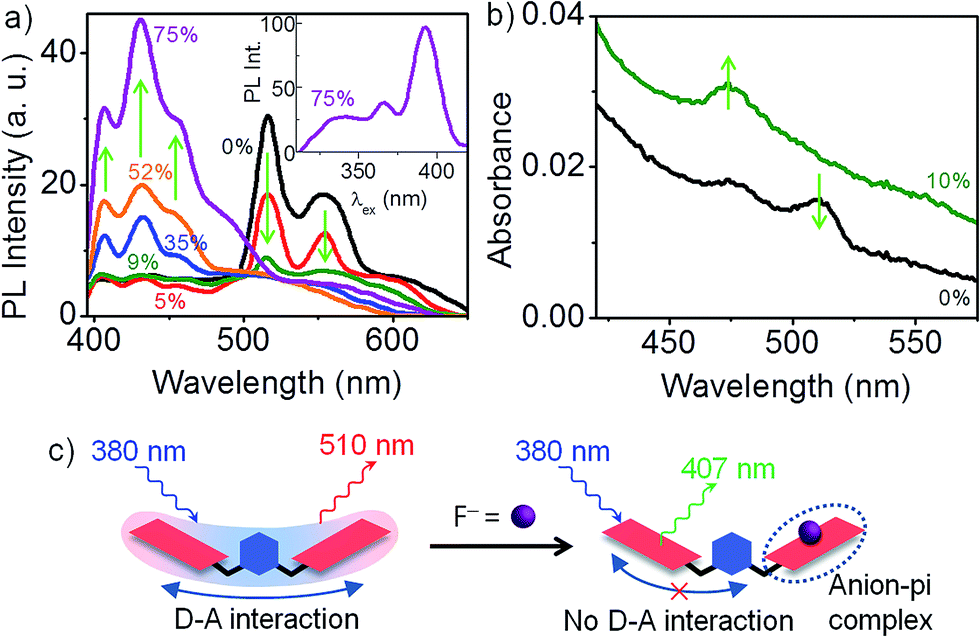 | ||
| Fig. 2 F− binding with dimer 1 (a) change in PL spectra (20 μM in CH2Cl2, λex = 380 nm) with incremental addition of tetra(n-butyl)ammonium fluoride shows a rapid decline in 510 nm PL and a concomitant rise in free NDI emission at 430 nm. F− mole percentage is noted. Inset: PLE spectrum recorded for λPL = 430 nm shows that the PL is indeed from free NDI. (b) Absorption spectrum shows that D–A absorbance is completely replaced by a 470 nm absorbance corresponding to 1:1 NDI–F− complex. (c) Schematic summarizes the competition between F− and the aromatic ring donors. | ||
The results presented so far illustrates the nature of D–A interaction between the xylylene bridge and a pair of non-interacting NDI chromophores. We now explore the fate of D–A interaction as the non-interacting NDI units of 1 fold into a strongly interacting pi-stack. Intramolecular pi-stacking in 1 was carried out in a mixture of good (CH2Cl2) and bad (methylcyclohexane – MCH) solvents at very low (10−5 M) concentration. Fig. 3a presents the absorption spectrum of 1 in CH2Cl2/MCH solvent mixture. At higher MCH compositions, there is an overall reduction of light extinction and a reversal in the relative intensities of 0–0 and 0–1 vibronic features. These changes are consistent with an H-type aggregation.15 Further, appearance of a new shoulder at 400 nm indicates that the transition to the lower exciton state of the H-type aggregate is also partially allowed. Absorption spectrum of 1 in 90% MCH confirms that the dimer is folded into its aggregated form. Persistence of the characteristic 510 nm PL emission in aggregated 1 clearly suggests that an excitonic coupling between the NDI units do not disrupt the 1:2 D–A interaction in 1 (inset to Fig. 3b). Direct excitation of the intramolecular H-type aggregate at 380 nm (Fig. 3b) leads to a composite PL spectrum, where at least two distinct emissive species can be identified. In addition to the 510 nm PL from the D–A species, a broad PL emission appears at 570 nm, which is presumably from an excimer (NDI*–NDI) species.4 Dual emission suggests that the initial photoexcitated state of the aggregated dimer can relax through two competing pathways with comparable rate constants, leading to either the excimer, or the D–A state.
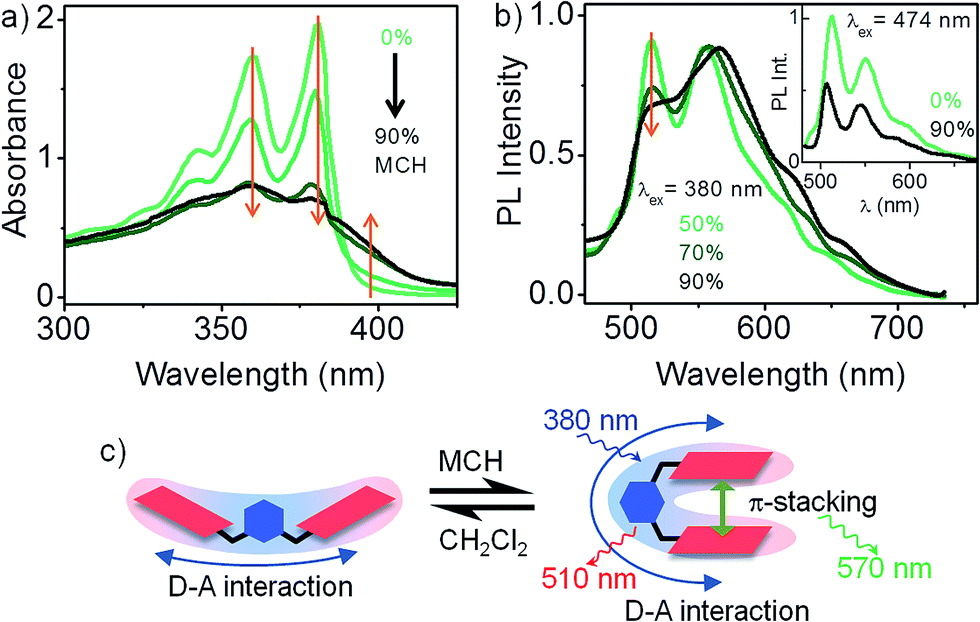 | ||
| Fig. 3 Effect of intramolecular pi-stacking on the D–A interaction. (a) Absorption spectra of 1 in CH2Cl2/MCH solvent mixture. Above 50% MCH composition, 1 folds into an intramolecular H-type aggregate, (b) normalized PL spectra (λex = 380 nm) of 1 in CH2Cl2/MCH solvent mixture shows dual emission: PL from NDI excimer at 570 nm in addition to a 510 nm PL form the D–A state. Inset: 474 nm excitation gives rise to the characteristic 510 nm PL from the D–A state in fully aggregated 1. (c) Schematic summarizes the evolution of PL as 1 folds into an intramolecular H-type aggregate. | ||
Conclusions
In summary, we report a unique intramolecular D–A interaction in a pair of regioisomeric covalently-bridged NDI dimers, 1 and 2. Though the D–A interaction is characterized by a weak absorbance, it exclusively dominates the photoluminescence properties of the dimers. Vibronically resolved PL and PLE spectra exhibiting mirror-image symmetry, a negligible stokes shift and a weak solvatochromism, points to a partial charge-transfer character of the D–A state. Using F− binding studies, we established that the D–A interaction involves one donor (xylylene ring) and a pair of non-interacting acceptor (NDI) units. This is further supported by the absence of a similar D–A species in 3. Finally, we have demonstrated that the D–A interaction persists even in the presence of a strong excitonic interaction between the NDI units. The significant of this finding should be seen in the context of supramolecular nanostructures of those donor–acceptor (D–A) type molecules, where only one part of the molecule (say acceptor) aggregates to create the nanostructure. We have shown that the interaction that operates between the donor and the acceptor units at the molecular level, continues to be present between the donor and an “aggregate of acceptors”.Acknowledgements
The authors gratefully acknowledge IISER Kolkata and Department of Science and Technology (DST), India (Project: EMR/2014/000223) for financial support. SS acknowledges UGC for scholarship.Notes and references
- M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910 CrossRef CAS PubMed; A. Saeki, Y. Koizumi, T. Aida and S. Seki, Acc. Chem. Res., 2012, 45, 1193 CrossRef PubMed.
- D. Chaudhuri, D. Li, Y. Che, E. Shafran, J. M. Gerton, L. Zang and J. M. Lupton, Nano Lett., 2011, 11, 488 CrossRef CAS PubMed; A. T. Haedler, K. Kreger, A. Isaac, B. Wittmann, M. Kivala, N. Hammer, J. Koehler, H.-W. Schmidt and R. Hildner, Nature, 2015, 523, 196 CrossRef PubMed.
- Z. Chen, V. Stepanenko, V. Dehm, P. Prins, L. D. Siebbeles, J. Seibt, P. Marquetand, V. Engel and F. Würthner, Chem.–Eur. J., 2007, 13, 436 CrossRef CAS PubMed; I. A. Howard, F. Laquai, P. E. Keivanidis, R. H. Friend and N. C. Greenham, J. Phys. Chem. C, 2009, 113, 21225 Search PubMed.
- B. Fimmel, M. Son, Y. M. Sung, M. Grüne, B. Engels, D. Kim and F. Würthner, Chem.–Eur. J., 2015, 21, 615 CrossRef CAS PubMed; Y. Wu, M. Frasconi, D. M. Gardner, P. R. McGonigal, S. T. Schneebeli, M. R. Wasielewski and J. F. Stoddart, Angew. Chem., Int. Ed., 2014, 53, 9476 CrossRef PubMed; R. J. Lindquist, K. M. Lefler, K. E. Brown, S. M. Dyar, E. A. Margulies, R. M. Young and M. R. Wasielewski, J. Am. Chem. Soc., 2014, 136, 14912 CrossRef PubMed; K. E. Brown, W. A. Salamant, L. E. Shoer, R. M. Young and M. R. Wasielewski, J. Phys. Chem. Lett., 2014, 5, 2588 CrossRef PubMed; J. M. Giaimo, J. V. Lockard, L. E. Sinks, A. M. Scott, T. M. Wilson and M. R. Wasielewski, J. Phys. Chem. A, 2008, 112, 2322 CrossRef PubMed.
- L. Zang, Acc. Chem. Res., 2015, 48, 2705 CrossRef CAS PubMed.
- T. Earmme, Y. J. Hwang, N. M. Murari, S. Subramaniyan and S. A. Jenekhe, J. Am. Chem. Soc., 2013, 135, 14960 CrossRef CAS PubMed; X. Guo, F. S. Kim, M. J. Seger, S. A. Jenekhe and M. D. Watson, Chem. Mater., 2012, 24, 1434 CrossRef.
- E. Ahmed, G. Ren, F. S. Kim, E. C. Hollenbeck and S. A. Jenekhe, Chem. Mater., 2011, 23, 4563 CrossRef CAS; M. R. Molla, A. Das and S. Ghosh, Chem.–Eur. J., 2010, 16, 10084 CrossRef PubMed; O. Yushchenko, D. Villamaina, N. Sakai, S. Matile and E. Vauthey, J. Phys. Chem. C, 2015, 119, 14999 Search PubMed.
- C. Kulkarni, G. Periyasamy, S. Balasubramanian and S. J. George, Phys. Chem. Chem. Phys., 2014, 16, 14661 RSC; S. Basak, S. Bhattacharya, A. Datta and A. Banerjee, Chem.–Eur. J., 2014, 20, 5721 CrossRef CAS PubMed.
- Y. Takashima, V. Martinez-Martinez, S. Furukawa, M. Kondo, S. Shimomura, H. Uehara, M. Nakahama, K. Sugimoto and S. Kitagawa, Nat. Commun., 2011, 2, 168 CrossRef PubMed.
- S. V. Bhosale, C. H. Jani and S. J. Langford, Chem. Soc. Rev., 2008, 37, 331 RSC.
- T. Ono, M. Sugimoto and Y. Hisaeda, J. Am. Chem. Soc., 2015, 137, 9519 CrossRef CAS PubMed.
- T. D. M. Bell, S. Yap, C. H. Jani, S. V. Bhosale, J. Hofkens, F. C. DeSchryver, S. J. Langford and K. P. Ghiggino, Chem.–Asian. J., 2009, 4, 1542 CrossRef CAS PubMed.
- H. Shao, T. Nguyen, N. C. Romano, D. A. Modarelli and J. R. Parquette, J. Am. Chem. Soc., 2009, 131, 16374 CrossRef CAS PubMed; H. Shao, J. Seifert, N. C. Romano, M. Gao, J. J. Helmus, C. P. Jaroniec, D. A. Modarelli and J. R. Parquette, Angew. Chem., Int. Ed., 2010, 49, 7688 CrossRef PubMed.
- S. Guha and S. Saha, J. Am. Chem. Soc., 2010, 132, 17674 CrossRef CAS PubMed; R. E. Dawson, A. Hennig, D. P. Weimann, D. Emery, V. Ravikumar, J. Montenegro, T. Takeuchi, S. Gabutti, M. Mayor, J. Mareda, C. A. Schalley and S. Matile, Nat. Chem., 2010, 2, 533 CrossRef PubMed.
- F. C. Spano, Acc. Chem. Res., 2010, 43, 429 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization, experimental methods, PL quantum efficiency. See DOI: 10.1039/c6ra02133f |
| ‡ Both absorption and PL spectra of 1 and 2 in CH2Cl2 remains the same, even at high (∼2 mM) concentrations (see ESI†). |
| This journal is © The Royal Society of Chemistry 2016 |