DOI:
10.1039/C6RA01784C
(Paper)
RSC Adv., 2016,
6, 34342-34349
Graphene based nanoassembly for simultaneous detection and degradation of harmful organic contaminants from aqueous solution†
Received
20th January 2016
, Accepted 16th March 2016
First published on 17th March 2016
Abstract
Graphene based nanoassemblies that can simultaneously detect and degrade harmful organic contaminants from water are important for conquering the risk of hazardous chemicals. A unique semiconductor–metal–graphene nanosheet assembly design has been successfully fabricated by a wet chemical method. Such a nanoassembly arrangement is made up of ZnO–Ag core–shell nanostructures and integrates on graphene nanosheets (GNSs). High-resolution scanning transmission electron microscopy reveals that ZnO–Ag core–shell nanostructures consist of a core ZnO nanosphere covered by a Ag shell and are chemically decorated on the GNS surface. This ZnO–Ag–GNS nanoassembly is SERS active and hence able to sense organic contaminants such as acridine orange dye. The sunlight-driven photocatalytic activity of the ZnO–Ag–GNS nanoassembly design demonstrates the tremendous enhancement in the detection and degradation of organic contaminants. The results in the present study show that the enhancement in the photocatalytic activity is attributed to the interfacial charge transfer effect in ZnO–Ag–GNS interfaces. This new design exhibits an efficient photocatalytic degradation of organic dye under sunlight irradiation. This could have great potential for a high sensing, stable and reusable material for energy and environmental cleaning applications.
1. Introduction
Sensing and destroying of harmful organic contaminants in bodies of water remain a major problem in the ecological system. Toxic organic dye effluents are released from various industries like tanneries, textiles, etc., and contain nitroaromatic dyes used as colouring agents, finding their way into rivers. These hazardous contaminants will destroy the ecosystem in the near future. The photocatalytic performances of semiconductor based nanoassemblies are helpful in the remediation of hazardous organic contaminants from bodies of water.1a–c
Under excitation with energized photons, electrons and holes in a semiconductor need to separate, allowing them to initiate degradation of organic dyes at the interface. ZnO, a wide band gap n-type semiconducting nanomaterial, is a widely studied system for photocatalytic applications.2a,b For a photocatalytic reaction process, the reaction molecules receive electrons and holes from surface sites for further reduction reactions. In competition with the charge carriers (i.e., electrons and holes), most of them are recombined due to wide band gap energy and instability in the higher state, which is a crucial factor that limits the photocatalytic activity by low quantum efficiency.3a–c In order to avoid the rapid recombination rate of an electron–hole pair, a widely reported approach is to incorporate metal nanomaterials on a n-type semiconductor.4–8 The photogenerated electrons transfer to the metal nanomaterial across a junction and become trapped owing to the Schottky barrier whereas the holes generously spread to the semiconductor surface.
There are two factors that cause the reduction in the electron–hole pair recombination rate: (i) the appearance of defects during the formation of interface engineering in the hybrid nanostructures7a–c and (ii) some electron rich co-catalysts with semiconducting metal oxides are incorporated to form a hetero-junction between the host semiconductor and the co-catalyst that generates a local electric field, which separates the electron–hole pairs completely.11,12 However, there are certain countable limitations that occur owing to the lack of bulk-to-surface sites in the created interfaces that reduce the exposed metal–semiconductor surfaces and thus make it complicated for the reaction molecules to lift up the photo-induced charge carriers for reduction reactions on the surface.9,10
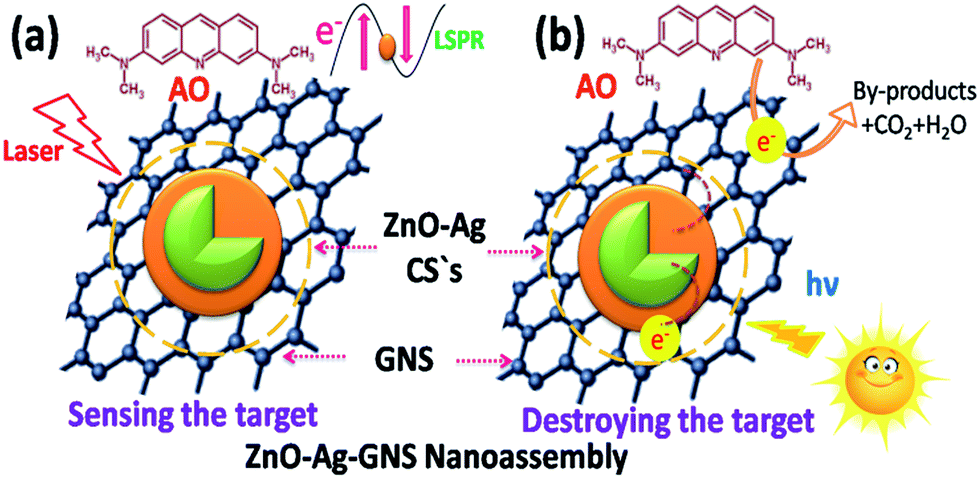
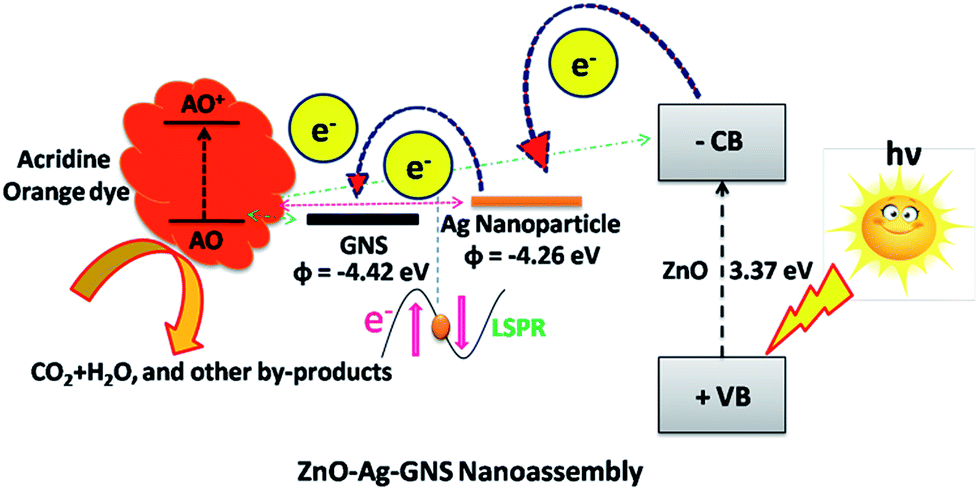
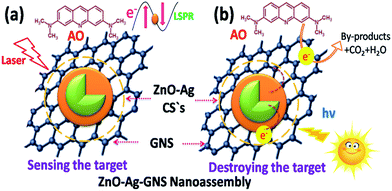
Subsequently, we decided to adopt a new materials design to conquer this undesirable condition. This new nanoassembly is made by integrating a graphene nanosheet, which is a well identified two-dimensional material with excellent physio-chemical properties and very high electron mobility, into a fabricated n-type semiconductor (ZnO)–metal (Ag) core–shell nanostructure. Interestingly, Ag nanoparticles showing a localized surface plasmon resonance effect (LSPR) improve Raman scattering of organic contaminants and thus facilitate the sensing through surface enhanced Raman spectroscopy (SERS).11–13 As the graphene nanosheet is in close contact with the metal nanomaterial in this assembly, it is anticipated that the electrons gathered on the conduction band of the semiconductor can transfer to the graphene nanosheets through a metal–graphene pathway and are entirely taken up by oxidized molecules and, simultaneously, the holes reside on the semiconductor for reduction reactions.14–20 The adaptability of this new nanoassembly is superior than others recently reported in the literature, because of its capability of both sensing and rapid photodegradation of organic contaminants in aqueous medium.21a–e The significance of the each component in the ZnO–Ag–GNS nanoassembly is portrayed in Fig. 1.
 |
| | Fig. 1 (a) GNSs and Ag Nps work to enable detection of organic contaminants in an aqueous solution and (b) photoinduced charge carriers from the semiconductor can transfer to the graphene nanosheets through a metal–graphene pathway and entirely diffused the target molecules. | |
In this work, we demonstrate a unique semiconductor–metal–graphene nanoassembly design, which can perfectly overcome the above mentioned limitations and thus enhances the photocatalytic activity by LSPR and interfacial charge transfer effects. This unique nanoassembly is versatile and can simultaneously detect and degrade the organic contaminants dispersed in the aqueous solution under natural sunlight irradiation.
2. Experimental section
2.1 Materials
All the chemicals used in this work were of analytical grade and were used without any further purification. Graphite (325 mesh, Alfa Aesar), potassium permanganate (Sigma-Aldrich), hydrogen peroxide (30 wt%, Sigma-Aldrich), sulfuric acid (Rankem Chemicals), hydrochloric acid (Rankem Chemicals), zinc acetate dihydrate (Fischer Chemicals), oxalic acid (Fischer Chemicals), silver nitrate (SRL Ltd.), ethanol (Hayman limited), and acridine orange dye (Fischer Chemicals) were used.
2.2 Synthesis of ZnO nanomaterials
In a typical experiment, 1.0 M of zinc acetate [Zn(CH3COO)2·2H2O] was dissolved in double-distilled water (200 mL) at ∼5 °C and stirred for 30 minutes. 1.5 M of oxalic acid (H2C2O4·2H2O) dissolved in double-distilled water (200 mL) at ∼5 °C was slowly added to the cold solution of zinc acetate. The mixture was further stirred for 2 hours at low temperature till the solution gradually turned into a white precipitate. The precipitate was then filtered and washed with ethanol and allowed to dry at 80 °C for 2 hours and that ended up in zinc oxalate formation. Zinc oxalate was further calcined at 400 °C for 2 hours to obtain pure ZnO nanomaterial.
2.3 Preparation of ZnO–Ag core–shell nanostructures
Experimentally, a facile wet chemical approach has been developed to fabricate the ZnO–Ag core–shell nanostructures with different Ag shell thickness. In detail the experimental procedure is as follows: a given amount of silver nitrate (1 mM, 4 mM and 7 mM) and 100 mg of zinc oxide were dispersed in 100 mL of ethanol under vigorous magnetic stirring. Subsequently, the suspension was aged at room temperature for 2 hours and finally, it was dried isothermally at 80 °C for 12 hours and then the residue was transferred into the furnace at various temperatures for 2 hours to crystallize the materials. The thickness of the Ag shell could be varied by changing the concentration of the silver nitrate precursor as 1 mM, 4 mM and 7 mM and was named as ZnO–Ag-1, ZnO–Ag-2 and ZnO–Ag-3 respectively.
2.4 Synthesis of the ZnO–Ag–GNS nanoassembly
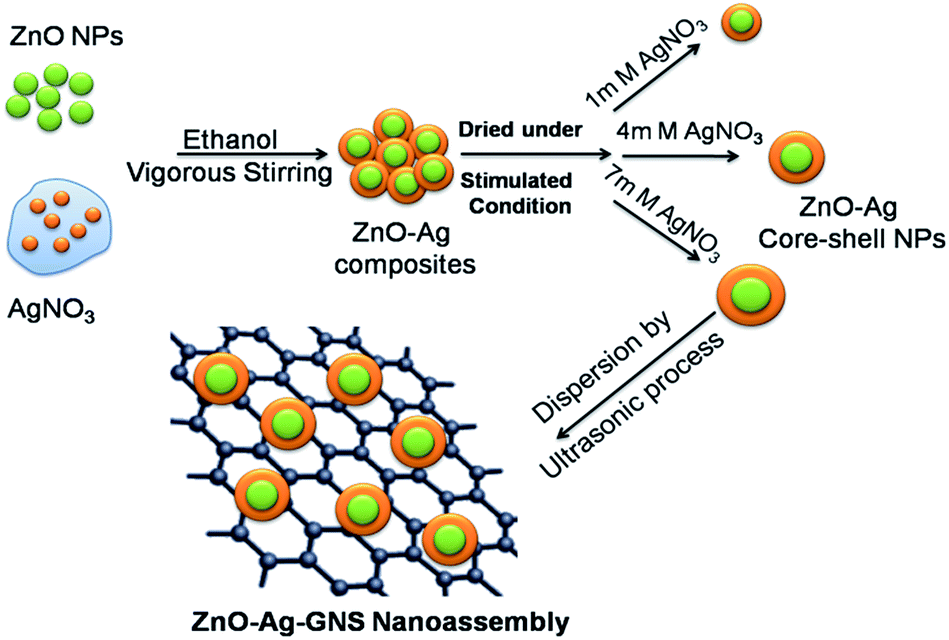
The synthesis procedure for the ZnO–Ag–GNS nanoassembly is schematically explained in Fig. 2. The as-synthesized GNSs (100 mg) were dispersed in double distilled water (100 mL) using an ultrasonicator to form a colloidal suspension. The GNS solution was added to a higher silver nitrate concentration of the ZnO–Ag core–shell (ZnO–Ag-3) suspension (100 mg of material is dispersed in 100 mL double distilled water). The obtained precipitate was stirred vigorously and separated by centrifugation at 4000 rpm. The resulting product was washed with ethanol and dried in a vacuum oven at 100 °C for 24 hours to get a ZnO–Ag–GNS nanoassembly.
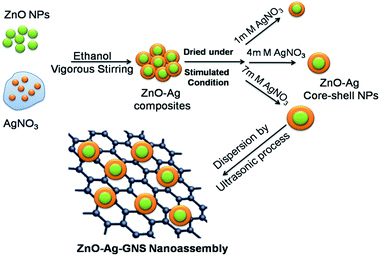 |
| | Fig. 2 Schematic illustration of ZnO–Ag–GNS nanoassembly synthesized by a wet chemical process. | |
2.5 Characterization
X-ray diffraction (XRD) data were collected with a PANalytical X-ray diffractometer using Cu Kα (λ = 1.54 Å) radiation. The samples were scanned in the 2θ range of 10° to 80°. All of the peaks were assigned and compared with data from the Joint Committee on Powder Diffraction Standards (JCPDS) database. XPS measurements were obtained using ESCA+, an Omicron Nanotechnology ESCA probe spectrometer with monochromatized Al Kα X-rays (energy: 1486.6 eV). The X-ray power applied was 300 W. The pass energy was 50 eV for survey scans and 20 eV for specific regions. The sample solution was spotted on a molybdenum sample plate and dried in a vacuum. Spectra in the required binding energy range were collected, and an average spectrum was taken. Each spectrum was confirmed after scanning eight times for confirmation (except the survey scan, which was scanned only once). While taking the spectra, the scan steps per second were fixed for all of the narrow scans. Beam-induced damage of the sample was reduced by adjusting the X-ray flux. The base pressure of the instrument was 5.0 × 10−10 mB. The binding energy was calibrated with respect to the adventitious C 1s feature at 284.6 eV. Most of the spectra were deconvoluted to their component peaks, using the software CASA-XPS. High-resolution scanning transmission electron microscopy (HRSTEM) of the samples was carried out using a FEI Tecnai G2 S-twin instrument with a UHR pole piece. HRSTEM samples were prepared by drop-casting two or three drops of the sample-dispersed ethanol solution to carbon-coated copper grids and allowed to dry at room temperature overnight. The photoluminescence (PL) spectrum was recorded using Perkin Elmer MPF-44B equipment under (325 nm) excitation.
2.6 Photocatalytic activity
The photocatalytic activity of the ZnO–Ag–GNS nanoassembly was evaluated by the degradation of acridine orange (AO) under solar light. In a typical process, 150 mL of a 3.0 × 10−5 M, 3.5 × 10−5 M or 4.0 × 10−5 M concentration of AO dye solution and 50 mg of ZnO–Ag–GNS nanostructures were stirred for about 1 hour. Before exposure to illumination, the suspensions were stirred in the dark for 30 minutes to ensure the establishment of adsorption/desorption equilibrium of AO on the sample surfaces. Consequently, the suspension was irradiated with direct sunlight and this was carried out for all the experiments under similar conditions on sunny days in Chennai city (geographical location 13.04° N and 80.17° E on the southern-east coast of India), between 12:00 p.m. and 3:00 p.m. (outside temperature, 29 °C to 31 °C). The measured illuminance power of the direct sunlight at a given interval of time was around 110![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 lux to 120000 lux obtained by using the Digital illuminance meter (TES Electrical Electronic Corp. Taiwan). At a given time interval of irradiation, 5 mL of the suspension was withdrawn and subsequently centrifuged at a rate of 3000 rpm for 5 minutes. UV-Vis absorption spectra of the supernatant were then measured using a UV-visible spectrophotometer.
000 lux to 120000 lux obtained by using the Digital illuminance meter (TES Electrical Electronic Corp. Taiwan). At a given time interval of irradiation, 5 mL of the suspension was withdrawn and subsequently centrifuged at a rate of 3000 rpm for 5 minutes. UV-Vis absorption spectra of the supernatant were then measured using a UV-visible spectrophotometer.
3. Results and discussion
3.1 Structural analysis
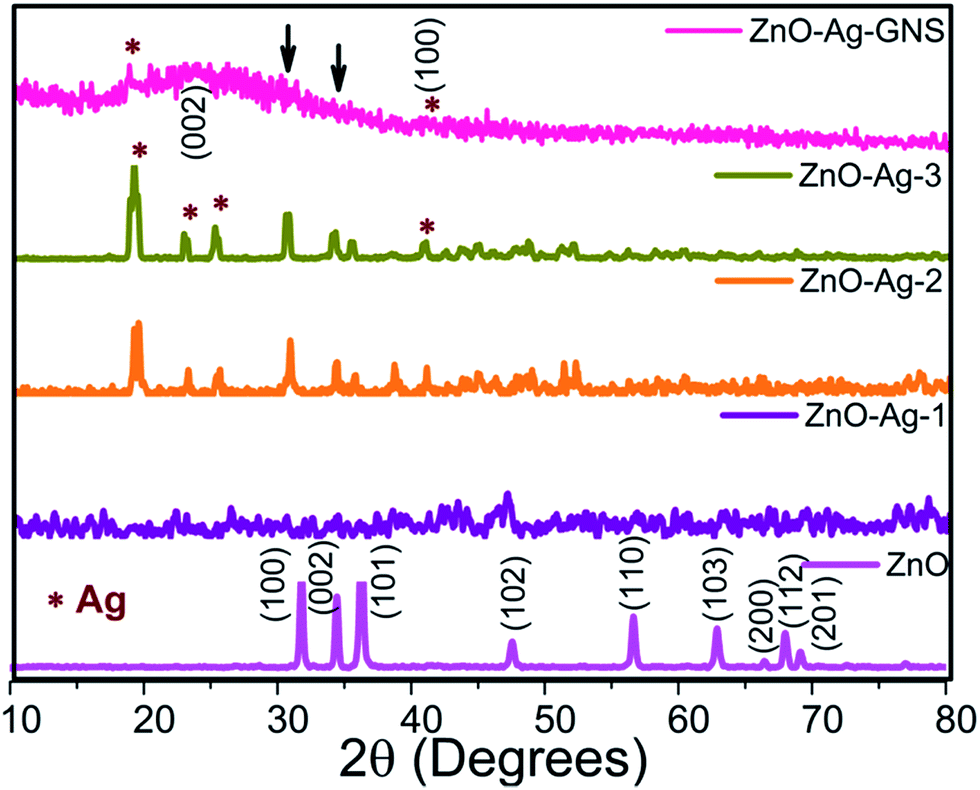
The crystalline structures, phase purity and textural features of the as-synthesized nanostructures were studied by XRD analysis. The XRD patterns of pure ZnO, ZnO–Ag core–shells and the ZnO–Ag–GNS stack design are shown in Fig. 3. All of the samples were crystalline in nature and of hexagonal wurtzite phase ZnO (JCPDS no. 36-1451). However, the ZnO–Ag core–shells exhibited additional Ag diffraction peaks located in the Fig. 3, which could be indexed to metallic silver with a face-centered cubic system (JCPDS no. 01-1167), signifying that the Ag nanomaterials were covered on the ZnO surface. By increasing the Ag concentration, the Ag peak intensity also increases accordingly.22,23 The XRD patterns of the ZnO–Ag–GNS stack show a broad peak at 25.7 and 41.16 degrees, corresponding to the (002) and (100) reflections. It is evident that the graphene oxide is significantly reduced to graphene nanomaterials.24a–c A very low quantity of the ZnO–Ag-3 core–shell nanostructure was integrated onto the GNS surfaces that can be evidently compared and analyzed with individual nanomaterials. However, the existence of metallic Ag in ZnO–Ag–GNS can be obviously elucidated by XPS analysis, as discussed in the next section.
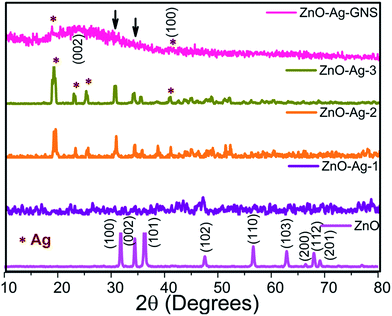 |
| | Fig. 3 XRD patterns of pure ZnO, different shell thickness of ZnO–Ag-1, ZnO–Ag-2, ZnO–Ag-3 and ZnO–Ag–GNS hybrid nanostructures. | |
3.2 Chemical state analysis
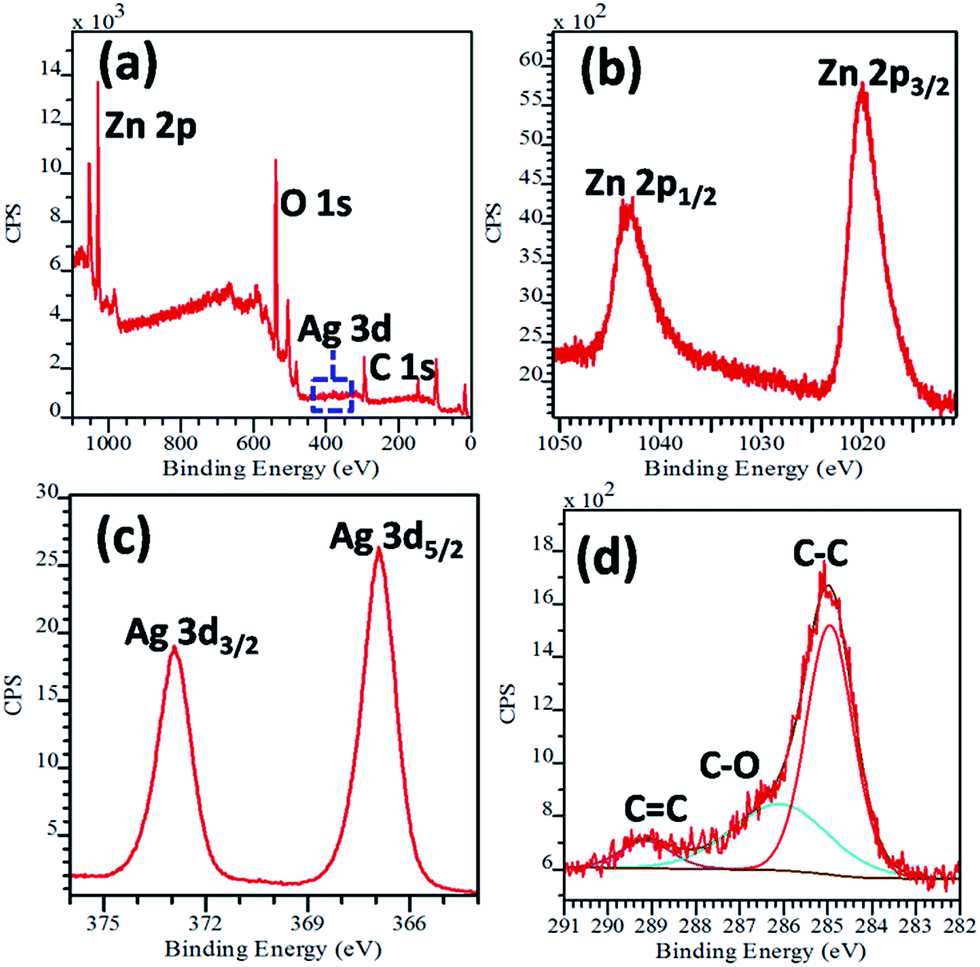
To obtain further essential information on the surface structure, chemical analysis and oxidation states of ions of the as-prepared nanostructures, XPS analysis was performed on the ZnO–Ag–GNS hybrid nanostructures. Fig. 4a exhibits the survey spectrum, which indicates that the sample contains Zn, O, Ag and C elements and evidently confirms that no other elements were identified. The peaks located at 1020.4 eV and 1043.4 eV correspond to Zn 2p3/2 and Zn 2p1/2, respectively, representing the existence of a Zn2+ state (in Fig. 4b).25 Fig. 4c shows the Ag 3d XPS spectra with two broad bands at 367.3 eV and 373.3 eV binding energies, which are ascribed to Ag 3d5/2 and Ag 3d3/2, respectively. No other peak corresponding to Ag2O or AgO was observed in the XPS spectra, which indicates that the Ag species coated on ZnO are in the form of a metallic state, not in Ag2O or AgO.26
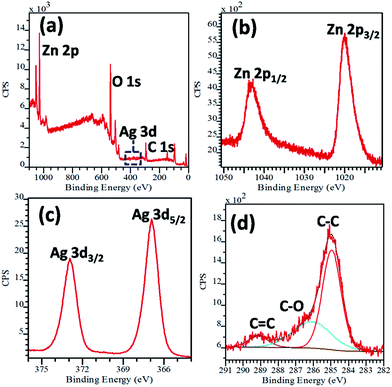 |
| | Fig. 4 (a) XPS survey spectra and (b) high resolution spectra of Zn 2p, (c) Ag 3d, and (d) C 1s regions of ZnO–Ag–GNS hybrid nanostructures. | |
The C 1s spectrum was deconvoluted into three peaks at 284.6 eV, 286.6 eV and 289.2 eV binding energies (in Fig. 4d). The peak at 284.6 eV is due to the C–C bond of graphene. The peak at 286.6 eV is attributed to the C–O bond, whereas the peak at 289.2 eV may possibly be assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond.26
C bond.26
3.3 Electron microscopy studies
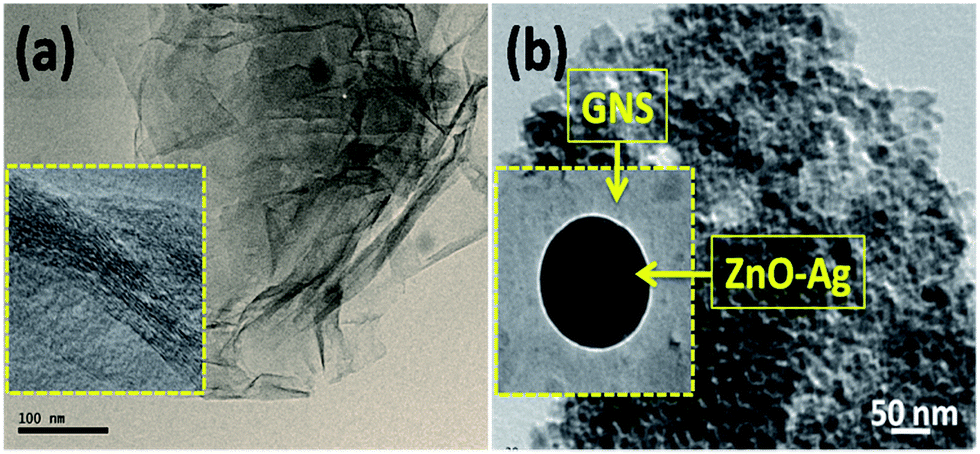
The electron microscopy images of the as-synthesized nanomaterials ZnO, ZnO–Ag-1, ZnO–Ag-2, ZnO–Ag-3 core–shell nanostructures and ZnO–Ag–GNS hybrid nanostructures, are given in Fig. 5 and 6. The surface morphology of the ZnO nanomaterial exhibits uniform spherical structure of diameters ranging from 20 nm to 25 nm, which is exhibited in Fig. 5a. To fabricate core–shell nanostructures with different shell thickness, the silver nitrate concentration was increased (1 mM, 4 mM and 7 mM) and coated onto ZnO under a stimulated reaction medium (in Fig. 5b–d). The thickness of the outer shell is in the range of ∼3 nm to 4 nm for ZnO–Ag-1, ∼4 nm to 6 nm for ZnO–Ag-2 and ∼6 nm to 8 nm for ZnO–Ag-3, respectively. The respective insets clearly show that the surfaces of ZnO were completely coated with the metallic Ag nanoshell of different thickness, which was obtained by increasing the concentration of the silver nitrate precursor, which is in good agreement with the XRD results.
 |
| | Fig. 5 Electron micrographs of (a) ZnO, (b) 1 mM Ag concentration of ZnO–Ag-1, (c) 4 mM concentration of ZnO–Ag-2 and (d) 7 mM concentration of ZnO–Ag-3 core–shell nanostructures. Respective insets show higher magnification images. | |
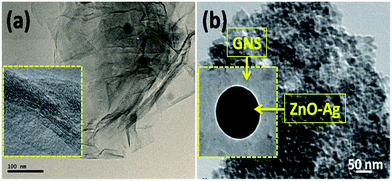 |
| | Fig. 6 Electron micrographs of (a) GNSs and (b) ZnO–Ag–GNS hybrid nanostructures. Respective insets show higher magnification images. | |
Fig. 6 evidently shows the formation of GNSs with an ultrathin wrinkled silk wave like structure. It is therefore apparent that GNSs definitely act as a two-dimensional support for the incorporation and growth of homogeneous core–shell nanostructures. After integration with a larger shell thickness of the ZnO–Ag core–shell nanostructure, the GNSs were completely decorated by crystalline ZnO–Ag core–shell nanostructures.
3.4 Detection of organic contaminants
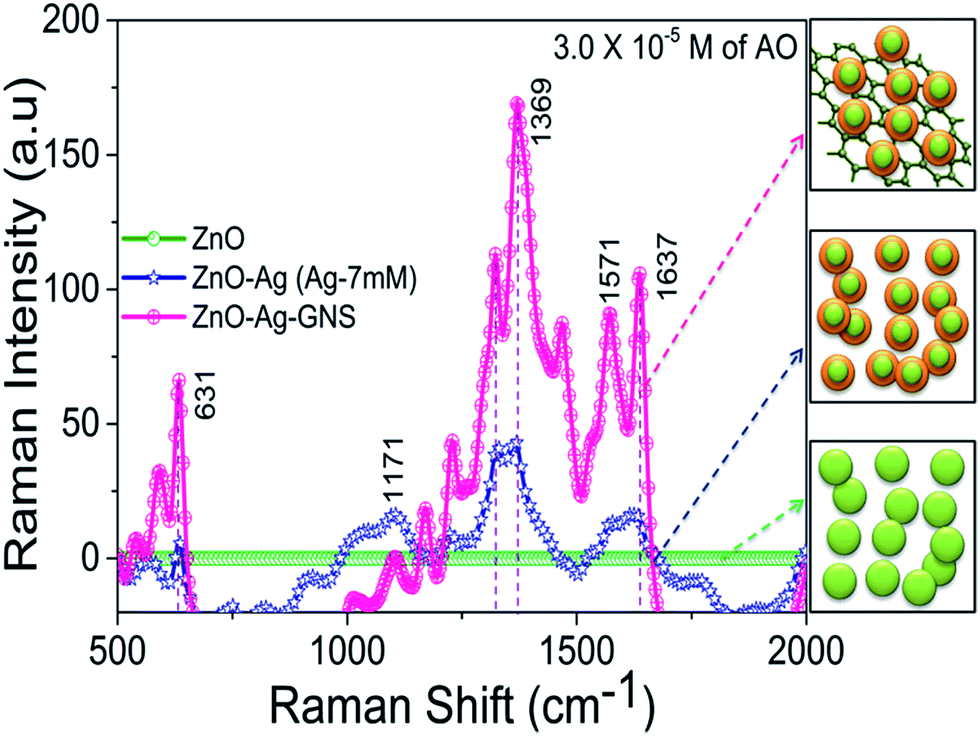
SERS is a versatile technique for the detection of organic molecules in an aqueous solution; it employs the interaction between the organic molecule (target) and the LSPR of Ag NPs to intensify the Raman signal of the organic molecule. Moreover, the GNSs in the ZnO–Ag–GNS nanoassembly can improve SERS signals due to quenching in the fluorescence effect along with their Raman-enhancing properties.27 Herein, we investigated the capability of this ZnO–Ag–GNS nanoassembly to perform as a chemical sensor by employing acridine orange as the target molecule. The ZnO–Ag–GNS nanoassembly was dispersed in aqueous solution of various concentrations of AO and SERS studies were carried out by using a 532 nm laser excitation source. Fig. 7 shows the SERS spectrum of ZnO, ZnO–Ag core–shells (7 mM concentration of Ag shell material) and the ZnO–Ag–GNS nanoassembly, which are in contact with 1 mL of a 3.0 × 10−5 M concentration of AO in aqueous solutions. A control experiment was carried out with AO in aqueous solution and with ZnO nanorods that showed no detectable Raman signal. However, the ZnO–Ag–GNS nanoassembly under the same conditions delivers a significant enhancement of the Raman signal of AO as the Ag shell layer was close in contact with GNSs, serving as SERS active sites. The prominent bands in the region between 500 cm−1 to 700 cm−1 were assigned to an out of plane ring and CH deformation modes. The weak band around 1171 cm−1 mainly arises due to the CH deformation and CN stretching vibration. Additionally, the bands between 1350 cm−1 to 1650 cm−1, corresponding to the CC and CN stretching vibrations, are also seen.28
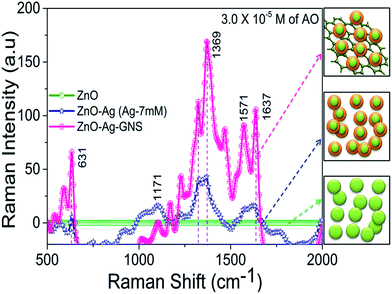 |
| | Fig. 7 SERS spectra of a 3.0 × 10−5 M concentration of AO in the presence of ZnO, ZnO–Ag core–shells and the ZnO–Ag–GNS nanoassembly. All the samples were kept in 1 mL of a 3.0 × 10−5 M concentration of AO in aqueous solution and allowed to immobilize for 30 min prior to the SERS experiment. | |
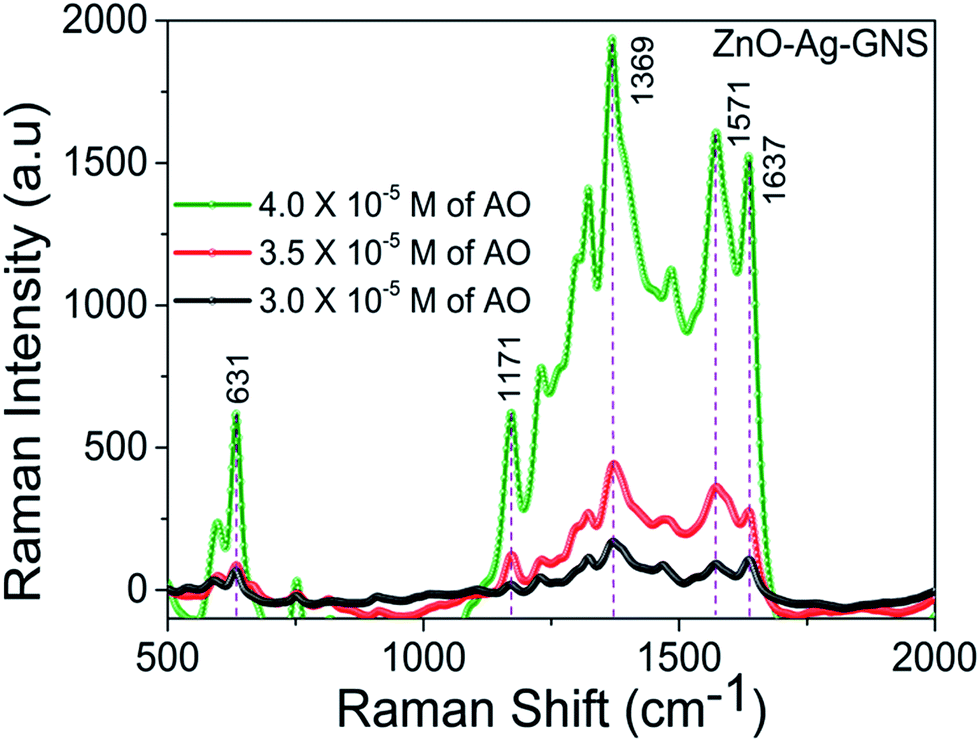
The ZnO–Ag–GNS nanoassembly sensor has the flexibility of detecting a wide range of organic molecules since the GNS is interfaced with the Ag shell layer, that can detect a low level of organic molecules from aqueous solution. Thus, the material construction of the ZnO–Ag–GNS nanoassembly provides a flexible route for detection of a wide range of organic molecules at lower concentrations. For comparison, the SERS response of the ZnO–Ag–GNS nanoassembly by varying AO concentration is shown in Fig. 8. The GNS not only gives a 2-D platform for the fabrication of a chemical sensor but also adsorbs organic molecules with higher chemical concentrations. Furthermore, the organic molecules interact with the GNS through van der Waals forces, which assist in the concentration of organic molecules onto the surface of the GNS. These interactions provide a means to attract and immobilize the organic molecules (pollutants) at the material interface and simultaneously degrades the pollutants under the exposure of natural sunlight.29,30 However, the degradation mechanism of organic pollutants using this material can be obviously elucidated by photocatalytic experiments, as discussed in the following sections.
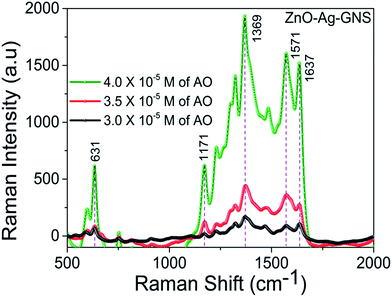 |
| | Fig. 8 SERS spectra of AO in the presence of the ZnO–Ag–GNS nanoassembly at AO concentrations of 3.0 × 10−5 M, 3.5 × 10−5 M and 4.0 × 10−5 M in aqueous solution. | |
3.5 Photocatalytic degradation of AO dye under direct sunlight irradiation
The photocatalytic activity of the as-prepared ZnO, ZnO–Ag core–shell, and ZnO–Ag–GNS hybrid nanostructures was studied by monitoring the degradation of a mutagenic organic pollutant (AO) under direct sunlight irradiation. The change in the absorption spectra of AO aqueous solutions showed the changes of its concentration. The initial concentration (C0), the residual concentration (C), and the degradation rate (D%) is illustrated by a mathematical expression as follows:| | |
D% = (C0 − C/C0) × 100%
| (1) |
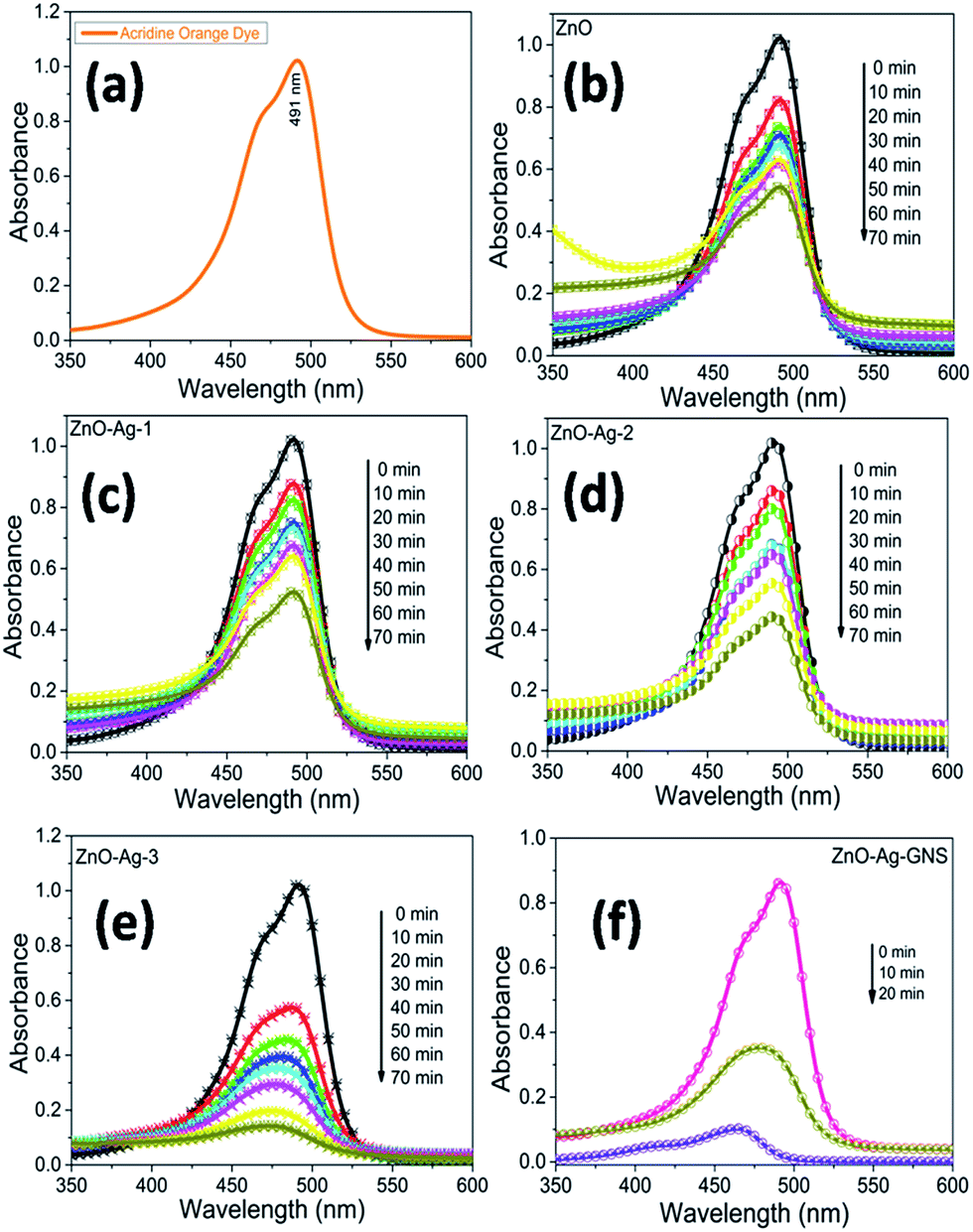
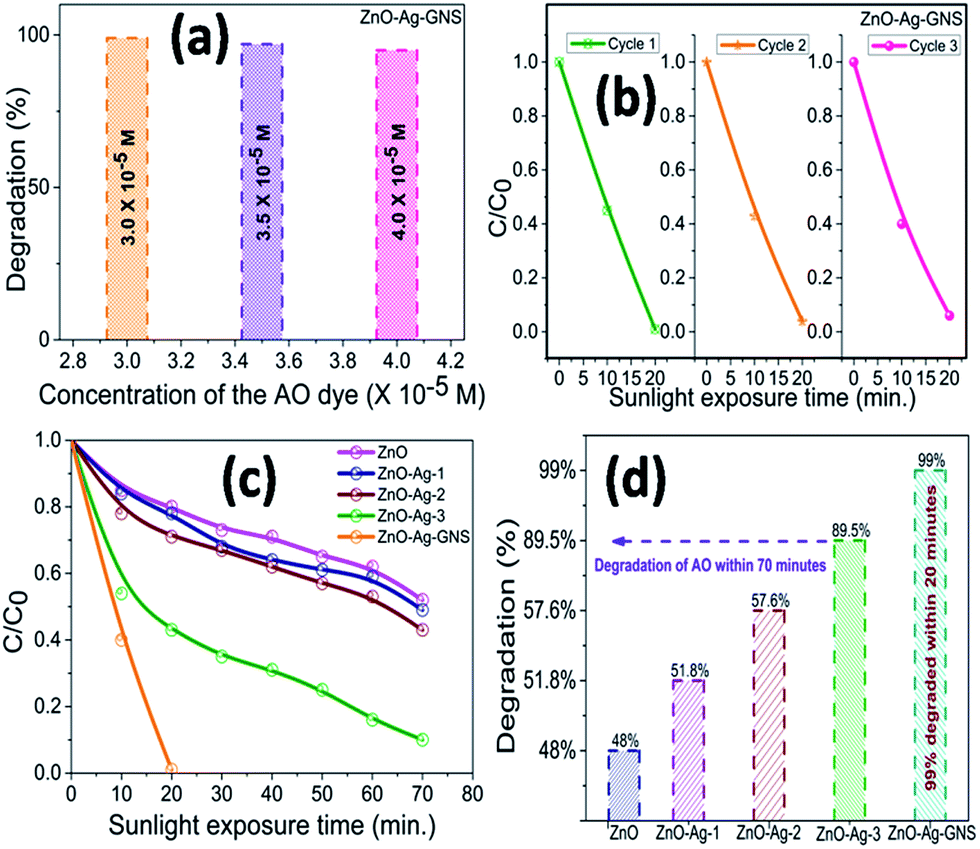
Fig. 9 shows the AO degradation plot versus time for ZnO, ZnO–Ag-1, ZnO–Ag-2, ZnO–Ag-3 core–shells, and ZnO–Ag–GNS nanoassembly photocatalysts, respectively. C/C0 spectra indicate that the 3.0 × 10−5 M concentration of AO dye was decomposed to about 48% for pure ZnO nanomaterial within 70 minutes, 51% for the ZnO–Ag-1 core–shell within 100 minutes, 57% for the ZnO–Ag-2 core–shell within 70 minutes, 89% for the ZnO–Ag-3 core–shell within 70 minutes and 99% for the ZnO–Ag–GNS hybrid nanostructures within 20 minutes (Fig. 10c and d) under direct sunlight irradiation. The ZnO–Ag–GNS nanoassembly showed higher photocatalytic efficiency than that of the pure and core–shell nanomaterials. This result was attributed to the GNS enhancement of the photocatalytic performance of ZnO–Ag core–shell nanomaterial under direct sunlight irradiation.
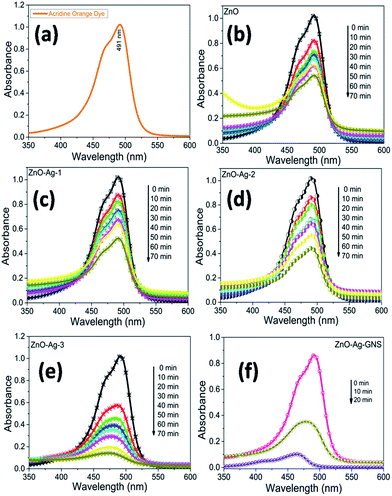 |
| | Fig. 9 Time-dependent photocatalytic degradation of AO dye under sunlight irradiation (a), and with ZnO (b), ZnO–Ag-1 (c), ZnO–Ag-2 (d), ZnO–Ag-3 (e) core–shells or ZnO–Ag–GNS hybrid nanostructures (f). | |
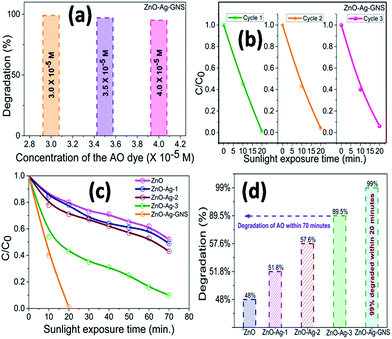 |
| | Fig. 10 Concentration dependent AO dye degradation under sunlight irradiation (a), stability and reusability studies (b) and C/C0 & degradation percentage of AO in the presence of as-prepared nanostructures (c & d). | |
Fig. 10(a) and (b) illustrate that the photocatalytic degradation process of AO over a ZnO–Ag–GNS nanoassembly photocatalyst under sunlight irradiation was effective. More importantly, by increasing the AO dye concentrations to 3.0 × 10−5 M, 3.5 × 10−5 M, and 4.0 × 10−5 M, and it was indicated that the ZnO–Ag–GNS nanoassembly photocatalyst could easily degrade the AO dye within 20 minutes of irradiation under direct sunlight and can be separated and reused by centrifugation. The stability of the nanoassembly photocatalysts was examined by three consecutive experimental cycles, which are very essential for the photocatalyst to be applied in environmental technology.
After three cycles of experiments, the ZnO–Ag–GNS photocatalyst still provides a high photodegradation rate under direct sunlight irradiation. It shows that the ZnO–Ag core–shells were decorated onto GNSs by electrostatic interaction which offers higher stability to the photocatalysts for multipurpose remediation processes. Hence, it could significantly promote its practical application to remove various harmful mutagenic organic pollutants from wastewater under direct sunlight irradiation.
3.6 Mechanism of enhanced photocatalytic activity under sunlight
From the above studies, the proposed mechanism of the efficient photocatalytic performance towards the degradation of an organic pollutant under sunlight could be credited mainly due to the LSPR effect and interfacial charge transfer process in this unique ZnO–Ag–GNS nanoassembly. We anticipate that the electrons gathered on the conduction band of the semiconductor can transfer to the graphene nanosheets through a metal-graphene pathway and be entirely taken up by oxidized molecules in the meantime, whereas the holes reside on the semiconductor for reduction reactions.10,26,31| | |
ZnO–Ag + hν → e− (Ag) + h+ (VB, ZnO)
| (2) |
| | |
GNS + e− (Ag) → e− (GNS) + ZnO
| (3) |
| | |
GNS + e− (AO) → e− (GNS) + AO
| (4) |
| | |
H2O + h+ (VB, ZnO) → ˙OH + H+
| (5) |
| | |
AO (ad) + ˙OH → CO2 + H2O
| (6) |
In the ZnO–Ag–GNS nanoassembly, the photogenerated electrons in ZnO can be moved to the conduction band and separate the holes in the valance band by absorbing solar spectrum due to the suitable narrow band gap of the ZnO–Ag core–shell structure. These photogenerated electrons can be transferred to GNSs and metallic Ag nanomaterials. On the other hand, photogenerated electrons cannot flow directly from AO to ZnO–Ag core–shell nanostructures (in Fig. 11), since there was a mismatch in their energy levels. A photo-excited electron from AO flows into CB of the ZnO core in the ZnO–Ag core–shell nanostructures via GNSs, where the photogenerated electrons react with oxygen molecules in the aqueous solution producing oxygen peroxide radicals. The positively charged holes break the hydroxide ion derived from the aqueous solution to form hydroxyl radicals.32–35 These oxygen peroxide radicals and hydroxyl radicals generated from the ZnO–Ag–GNS nanoassembly can mineralize and lead to the oxidative decomposition of the acridine orange dye to CO2, H2O, and other by-products. Surprisingly, the GNS has excellent electron mobility due to its two dimensional honeycomb structure, whereby the interfacial charge transport of photogenerated carriers could be achieved, and an effective charge separation is consequently proficient. Therefore, photodegradation is enhanced by excellent interfacial charge transport in the ZnO–Ag–GNS nanoassembly. Moreover, the influence of interface engineering could enhance the photocatalytic activity of ZnO–Ag–GNS hybrid nanostructures.
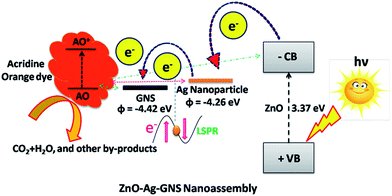 |
| | Fig. 11 Schematic illustration of a plausible mechanism involved in the charge transfer process from the different energy levels of ZnO–Ag–GNS for the photodegradation of acridine orange dye. | |
3.7 Photoluminescence studies
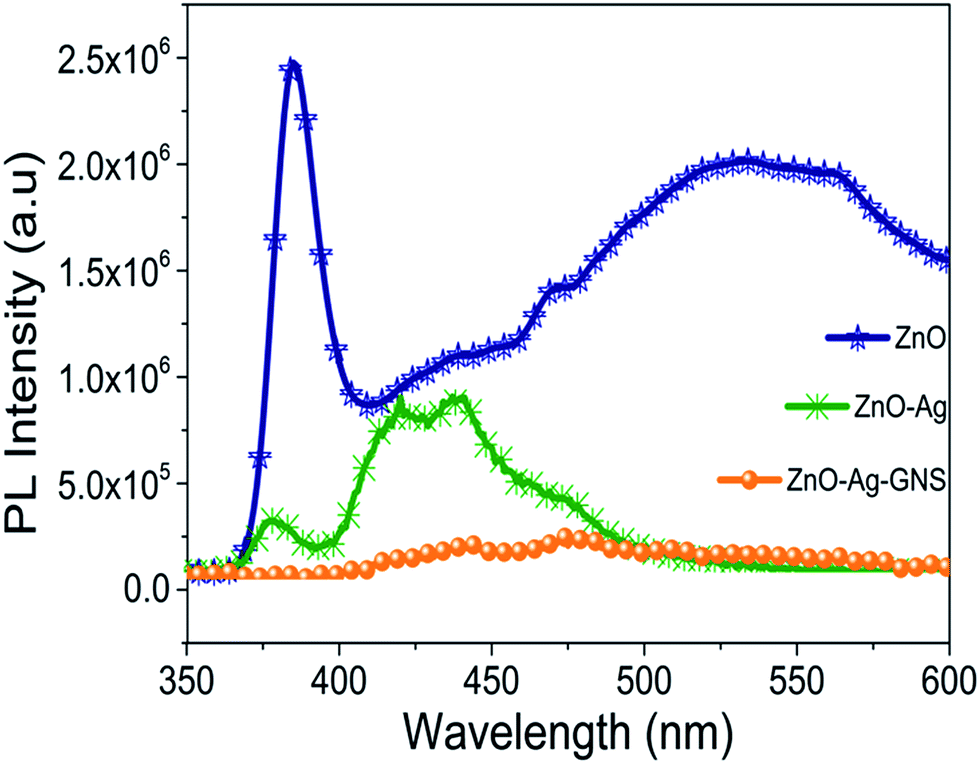
The charge separation property and recombination rate of photogenerated electron–holes can be identified by photoluminescence spectroscopy studies. The photogenerated electrons in the excited state can endure recombination with the holes, and results in irradiative release of energy in the form of fluorescence. Based upon the fluorescence emission characteristics of ZnO, ZnO–Ag and ZnO–Ag–GNS can be monitored by measuring room temperature photoluminescence spectra (in Fig. 12). It is shown that the peak at 385 nm shows a strong UV band and the weak green emission peak at 535 nm has been observed for pure ZnO nanomaterial due to its free excitonic emission near the band edge and local defect states respectively. In the PL spectra of the ZnO–Ag core–shell nanostructure compared with the pure ZnO nanomaterial, the intensity of the ZnO peaks decreases and shifts significantly due to the presence of the Ag layer.36,37 It is obvious that there is a heterojunction between the semiconductors that certainly reduces the defect during the core–shell formation process. A decrease in the PL emission intensity indicates better charge separation efficiency. Furthermore, emission intensity of the ZnO–Ag–GNS nanostructure peak decreased significantly, implying that the recombination rate of photogenerated charge carriers was effectively suppressed.9 As a result, the GNS is an excellent candidate for enhancing the photocatalytic activity in terms of prolonging the electron–hole pair lifetime, which drastically reduced the recombination rate of the electron–hole pair and accelerates the interfacial charge transfer process.
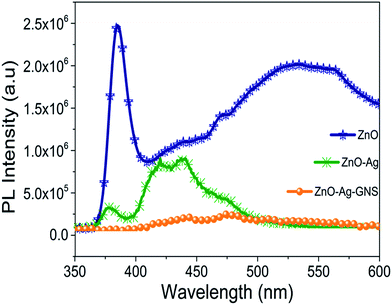 |
| | Fig. 12 Room temperature PL spectra of ZnO, ZnO–Ag core–shell and ZnO–Ag–GNS hybrid nanostructures. | |
4. Conclusions
In summary, the simultaneous detection and degradation of an organic contaminant by using a uniquely designed ZnO–Ag–GNS nanoassembly is demonstrated. Such a nanoassembly combines the ability to target organic molecules with SERS active sites and the photocatalytic activity provides a robust platform for multipurpose sensing and destroying applications. Moreover, this nanoassembly structure facilitates an efficient charge transfer process and the separation of the electron–hole. Consequently, the separation of electron–hole pairs is a significant process for all applications concerning semiconductor materials. It seems that this strategy may also be applicable to the synthesis of various hybrid nanostructures and also it recovers all the limitations and solutions in the photocatalytic field. We envisage that it will create a new development for designing hybrid nanomaterials for simultaneous sensing and photocatalytic environmental cleaning applications.
Acknowledgements
R. Ajay Rakkesh gratefully acknowledge the University of Madras for providing a NCNSNT fellowship (C2/AL/2011/388) supported by MHRD, India to carry out the research.
Notes and references
-
(a) M. Faisal, M. A. Tariq and M. Muneer, Dyes Pigm., 2007, 72, 233 CrossRef CAS;
(b) R. Arshad, S. Farooq, N. Iqbal and S. S. Ali, Lett. Appl. Microbiol., 2006, 42, 94 CrossRef CAS PubMed;
(c) R. Ajay Rakkesh and S. Balakumar, J. Nanosci. Nanotechnol., 2015, 15, 4316 CrossRef.
-
(a) L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735 CrossRef;
(b) Z. S. Wua, G. Zhoua, L. C.Yina, W. Rena, F. Lia and H. M. Chenga, Nano Energy, 2012, 1, 107 CrossRef.
-
(a) Z. Yin, S. Wu, X. Zhou, X. Huang, Q. Zhang, F. Boey and H. Zhang, Small, 2010, 6, 307 CrossRef CAS PubMed;
(b) R. Ajay Rakkesh and S. Balakumar, Processing and Applications of Ceramics, 2014, 8, 7 CrossRef;
(c) R. Ajay Rakkesh and S. Balakumar, J. Nanosci. Nanotechnol., 2013, 13, 370 CrossRef.
- R. K. Upadhyay, N. Soinb and S. S. Roy, RSC Adv., 2014, 4, 3823 RSC.
- F. Perreault, A. Fonseca de Faria and M. Elimelech, Chem. Soc. Rev., 2015, 44, 5861 RSC.
- E. Maggio and A. Triosi, J. Phys. Chem. C, 2013, 117, 196 Search PubMed.
-
(a) Y. T. Liang, B. K. Vijayan, K. A. Gray and M. C. Hersam, Nano Lett., 2011, 11, 2865 CrossRef CAS PubMed;
(b) R. Ajay Rakkesh, D. Durgalakshmi and S. Balakumar, J. Mater. Chem. C, 2014, 2, 6827 RSC;
(c) R. Ajay Rakkesh, D. Durgalakshmi and S. Balakumar, RSC Adv., 2015, 5, 18633 RSC.
- X. Zhang, Y. L. Chen, R. S. Liu and D. P. Tsai, Rep. Prog. Phys., 2013, 76, 401 CrossRef PubMed.
- M. Ahmad, E. Ahmed, Z. L. Hong, N. R. Khalid, W. Ahmed and A. Elhissi, J. Alloys Compd., 2013, 577, 717 CrossRef CAS.
- P. Dou, F. Tan, W. Wang, A. Sarreshteh, X. Qiao, X. Qiu and J. Chen, J. Photochem. Photobiol., A, 2015, 302, 17 CrossRef CAS.
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435 CrossRef CAS PubMed.
- M. G. Blaber, A. I. Hendry, J. M. Bingham, G. C. Schatz and R. P. Van Duyne, J. Phys. Chem. C, 2012, 116, 393 CAS.
- P. V. Kamat and D. Meisel, Curr. Opin. Colloid Interface Sci., 2002, 7, 282 CrossRef CAS.
- W. Tu, Y. Zhou, Q. Liu, S. Yan, S. Bao, X. Wang, M. Xiao and Z. Zou, Adv. Funct. Mater., 2013, 23, 1743 CrossRef CAS.
- S. Park, J. An, R. D. Piner, I. Jung, D. Yang, A. Velamakanni, S. T. Nguyen and R. S. Ruoff, Chem. Mater., 2008, 20, 6592 CrossRef CAS.
- B. Li and H. Cao, J. Mater. Chem., 2011, 21, 3346 RSC.
- M. Ahmad, E. Ahmed, Z. L. Hong, J. F. Xu, N. R. Khalid, A. Elhissi and W. Ahmed, Appl. Surf. Sci., 2013, 274, 273 CrossRef CAS.
- J. Liu, Y. Xue, Y. Gao, D. Yu, M. Durstock and L. Dai, Adv. Mater., 2012, 24, 2228 CrossRef CAS PubMed.
- I. V. Lightcap, T. H. Kosel and P. V. Kamat, Nano Lett., 2010, 10, 577 CrossRef CAS PubMed.
- L. Zhang, L. Du, X. Yu, S. Tan, X. Cai, P. Yang, Y. Gu and W. Mai, ACS Appl. Mater. Interfaces, 2014, 6, 3623 CAS.
-
(a) Y. Wen, H. Ding and Y. Shan, Nanoscale, 2011, 3, 4411 RSC;
(b) Y. Bu, Z. Chen and W. Li, RSC Adv., 2013, 3, 24118 RSC;
(c) F. Xu, Y. Yuan, D. Wu, M. Zhao, Z. Gao and K. Jiang, Mater. Res. Bull., 2013, 48, 2066 CrossRef CAS;
(d) D. H. Yoo, T. V. Cuong, V. H. Luan, N. T. Khoa, E. J. Kim, S. H. Hur and S. H. Hahn, J. Phys. Chem. C, 2012, 116, 7180 CrossRef CAS;
(e) J. Liu, L. Liu, X. Zhang and T. Li, New J. Chem., 2015, 39, 5272 RSC.
- R. S. Zeferino, M. B. Flores and U. Pal, J. Appl. Phys., 2011, 109, 014308 CrossRef.
- H. S. Kang, B. D. Ahn, J. H. Kim, G. H. Kim, S. H. Lim, H. W. Chang and S. Y. Lee, Appl. Phys. Lett., 2006, 88, 202108 CrossRef.
-
(a) L. Y. Meng and S. J. Park, Bull. Korean Chem. Soc., 2012, 33, 209 CrossRef CAS;
(b) R. Aladpoosh and M. Montazer, Carbohydr. Polym., 2016, 141, 116 CrossRef CAS PubMed;
(c) A. S. M. I. Uddin, K. W. Lee and G. S. Chung, Sens. Actuators, B, 2015, 216, 33 CrossRef.
- L. Sun, R. Shao, L. Tang and Z. Chen, J. Alloys Compd., 2013, 564, 55 CrossRef CAS.
- L. Sun, H. Yu and B. Fugetsu, J. Hazard. Mater., 2012, 203, 101 CrossRef PubMed.
- R. Alam, I. V. Lightcap, C. J. Karwacki and P. V. Kamat, ACS Nano, 2014, 8, 7272 CrossRef CAS PubMed.
- F. Zimmermann, B. Hossenfelder, J. C. Panitz and A. Wokaun, J. Phys. Chem., 1994, 98, 12796 CrossRef.
- H. Y. He, J. Klinowski, M. Forster and A. Lerf, Chem. Phys. Lett., 1998, 287, 53 CrossRef CAS.
- N. Jung, A. C. Crowther, N. Kim, P. Kim and L. Brus, ACS Nano, 2010, 4, 7005 CrossRef CAS PubMed.
- E. Lee, J. Hong, H. Kang and J. Jang, J. Hazard. Mater., 2012, 219, 13 CrossRef PubMed.
- D. Meisel and M. S. Matheson, Radiat. Res., 1983, 94, 604 Search PubMed.
- C. Harris and P. V. Kamat, ACS Nano, 2010, 4, 7321 CrossRef CAS PubMed.
- A. Takai and P. V. Kamat, ACS Nano, 2011, 5, 7369 CrossRef CAS PubMed.
- M. S Matheson, P. C. Lee, D. Meisel and E. Pelizzetti, J. Phys. Chem., 1983, 87, 394 CrossRef.
- F. Xu, Y. Yuan, D. Wu, M. Zhao, Z. Gao and K. Jiang, Mater. Res. Bull., 2013, 48, 2066 CrossRef CAS.
- W. Xie, Y. Li, W. Sun, J. Huang, H. Xie and X. Zhao, J. Photochem. Photobiol., A, 2010, 216, 149 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: UV-Vis DRS spectra of ZnO, ZnO–Ag core–shells and ZnO–Ag–GNS hybrid nanoassemblies. See DOI: 10.1039/c6ra01784c |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.