
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Confocal depth-resolved micro-X-ray absorption spectroscopy study of chemically strengthened boroaluminosilicate glasses
Pieter
Tack
 *a,
Stephen
Bauters
ab,
John C.
Mauro
c,
Morten M.
Smedskjaer
d,
Bart
Vekemans
a,
Dipanjan
Banerjee
b,
Wim
Bras
e and
Laszlo
Vincze
a
*a,
Stephen
Bauters
ab,
John C.
Mauro
c,
Morten M.
Smedskjaer
d,
Bart
Vekemans
a,
Dipanjan
Banerjee
b,
Wim
Bras
e and
Laszlo
Vincze
a
aDepartment of Analytical Chemistry, Ghent University, Krijgslaan 281 S12, 9000 Ghent, Belgium. E-mail: pieter.tack@ugent.be
bDutch-Belgian Beamline (DUBBLE), The European Synchrotron Radiation Facility (ESRF), 38043 Grenoble Cedex 9, France
cScience and Technology Division, Corning Incorporated, Corning, NY 14831, USA
dDepartment of Chemistry and Bioscience, Aalborg University, 9220 Aalborg, Denmark
eNetherlands Organisation for Scientific Research (NWO), The European Synchrotron (ESRF), 38043 Grenoble Cedex 9, France
First published on 26th February 2016
Abstract
It is crucial to understand the structural origins of macroscopic properties in silicate glasses for their high-tech applications. An example of such an application is chemically strengthened boroaluminosilicate glasses that are exposed to an ion exchange process during which alkali ions (e.g. Na+) are replaced by larger (e.g. K+) ions. Despite the empirically thorough understanding of this exchange process, much less is known about the fundamental physics of the process. Since Fe atoms are a suitable probe for monitoring chemical stress-induced changes in the local structure in the chemically strengthened glasses, a set of chemically strengthened boroaluminosilicate glasses containing 1 mol% Fe2O3 are here studied using depth-resolved confocal X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectroscopy. Information on the Fe oxidation state, coordination number, and bond distance as a function of the sample depth and glass composition is obtained. These new insights on chemical stress-induced changes will aid in the further development and improvement of such damage-resistant glasses.
1. Introduction
Understanding the structural origins of macroscopic properties in silicate glasses is crucial for their high-tech applications. The properties of glasses not only depend on the chemical composition, but also on the processing history. Thermal, pressure and chemical treatments can influence the macroscopic material properties to a large degree. For example, it is known that the coordination number (CN) of atoms or ions in materials increases through application of a sufficiently high pressure,1–4 a phenomenon that also applies to glassy materials.5–8 These modifications due to processing conditions are not necessarily uniform throughout the material, as part of the manufacturing process might be to induce depth dependent modifications. An example of this is the ion exchange process (IX), widely used in the glass industry.In this process, the to be processed glass object is placed in a molten inorganic salt bath (e.g. KNO3) at elevated temperatures whereupon smaller alkali ions (e.g. Na+) from the glass are replaced by larger ones (e.g. K+) from the molten salt bath. As a result, the outer layers of the glass have a different proportion of K2O and Na2O while the core of the material remains unchanged.
This exchange leads to formation of a compressive stress layer near the surface of the glass; it is well known and widely used by industry for production of chemically strengthened scratch and damage resistant glasses.9,10 As electronics devices are more and more portable, consumer demand dictates the devices not only having a physically pleasing appearance and functionality but also having excellent mechanical properties, including damage and scratch resistant surfaces. Some of the most successful examples are smartphones and tablets with cover glass post-treated by the IX process. The resulting high compressive stress of the material is the basis of the excellent mechanical properties of cover glasses.
Although the ion exchange process is empirically reasonably well understood,9,10 much less is known about the fundamental physics of the process. For instance, it is not known which chemical state the dopant elements are in as a function of diffusion depth in alkali-aluminosilicate glasses. According to molecular dynamics simulations of ion-exchanged glass, the CN of network formers is unchanged due to the IX process, whereas the invading alkali ions possess unique structures unattainable via standard melt-quenching processes.11–13 However, this has not yet been confirmed experimentally.
In this study, a set of boroaluminosilicate glasses, containing 1 mol% of Fe2O3 as dopant material, with varying SiO2 and Al2O3 concentrations are provided by Corning Inc. (NY, USA).14 Fe atoms are used here as a suitable probe for monitoring chemical stress-induced changes in CN, oxidation state, and bond distances in the glasses.15 Previous studies reported on the changes in CN of B, Al and the presence of non-bridging oxygen (NBO) in these glasses as a result of the different SiO2/Al2O3 ratios.16–19
Here we used depth-resolved X-ray absorption spectroscopy (XAS) to determine the chemical state and CN of Fe in the glasses. To monitor depth resolved differences in Fe state in the top 50 μm of the glass and in the bulk (∼100 μm depth), i.e., in compressive and tensile stress regions, respectively, polycapillary optics based confocal XAS measurements are performed, applying a rarely achieved 10 × 10 × 15 μm3 spatial resolution. The Fe–K X-ray absorption near-edge structure (XANES) spectroscopy pre-edge peak contains information on both the oxidation state and CN of the absorbing atom.20–26
Conventionally, information on the CN and local structure of the absorbing atom is obtained through extended X-ray absorption fine structure (EXAFS) spectroscopy experiments. Here, we opt not to perform EXAFS measurements for each sample at each depth due to the inherently longer acquisition time compared to XANES experiments (easily a factor 10 difference). Instead, EXAFS experiments are performed on a few selected samples at a depth near the surface (25 μm depth) and closer to the bulk (55 μm depth). This is done in order to compare with the XANES results and verify that the conclusions based upon the XANES results are valid even beyond the structural length scale for which XANES normally would render results.
A major objective of this approach is to minimize the local absorbed radiation dose since it has become clear in recent years that when glassy material is exposed to a too high local X-ray dose, the structural and chemical information obtained via X-ray based techniques might be compromised due to the generation of a high local concentration of photoelectrons.27–30 The radiation dose required to obtain valid XANES results is only a fraction from the dose required to obtain a complete EXAFS spectrum and therefore an assessment is made if the information rendered by XANES is sufficient or if complete EXAFS scans have to be made.
2. Experimental
Glass samples
A series of 10 boroaluminosilicate glass samples with varying SiO2 and Al2O3 concentration (see Table 1) and each containing approximately 1 mol% Fe2O3 were prepared by melt-quenching, as described in detail elsewhere.17 The glasses will be further referred to by their code as shown in the top row of Table 1. To induce a high compressive stress in the surface layers of the glasses, each glass sample was cut to a size of 2 × 2 × 0.1 cm3 and ion exchanged in a refined grade KNO3 molten salt bath for 4 hours at 410 °C. During this ion exchange process, Na+ ions are replaced by the larger K+ ions, generating a compressive stress in the glass surface, thereby improving its mechanical durability. Due to the nature of the diffusion-limited ion exchange process, more Na+ ions are replaced at the surface of the glass compared to the bulk of the glass.| Al0 | Al1 | Al2.5 | Al5 | Al7.5 | Al10 | Al12.5 | Al15 | Al17.5 | Al20 | |
|---|---|---|---|---|---|---|---|---|---|---|
| SiO2 | 79.4 | 78.9 | 77.4 | 74.7 | 71.8 | 68.9 | 67.1 | 64.1 | 62.3 | 61.1 |
| Al2O3 | 0.3 | 0.7 | 2.2 | 4.7 | 7.6 | 10.3 | 12.6 | 15.6 | 17.9 | 19.4 |
| Na2O | 14.6 | 14.5 | 14.6 | 14.6 | 14.7 | 14.8 | 14.3 | 14.3 | 13.7 | 13.6 |
| B2O3 | 4.9 | 4.9 | 4.9 | 5.0 | 4.9 | 5.0 | 5.0 | 5.0 | 5.1 | 5.0 |
| Fe2O3 | 0.9 | 0.9 | 0.9 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 0.9 | 0.9 |
XAS
Confocal XAS experiments were performed at BM26A, part of the Dutch-Belgian beam line (DUBBLE) at the ESRF.31 The beam line receives the radiation from a 0.40 T ESRF bending magnet. A double crystal Si(111) monochromator operating in a fixed-exit mode in combination with a collimating mirror allows an energy resolution ΔE/E of ∼1.7 × 10−4 at 9.659 keV. A vertically focusing mirror behind the monochromator allows vertical focusing and higher energy harmonic suppression.The primary intensity was determined with a gas-filled ion chamber and the transmitted intensity was determined with an identical device. The gas mixtures in both ionization chambers were adjusted to allow approximately 10% absorption in the first and about 70% absorption in the second ionization chamber at 7.212 keV. The photon flux was ∼6 × 109 photons per s at the sample position at the Fe K-edge energy (7112 eV). A 4 μm thick pure iron foil (Goodfellow SARL, Lille, France) was measured in transmission mode at regular time intervals during the experiment to correct for monochromator energy shifts by setting the first inflection point of the XANES profile to 7112 eV.
The incident X-ray beam was focused down to a 10(V) × 10(H) μm2 (FWHM) spot size at the sample position at the Fe–K edge energy using a polycapillary X-ray lens (XOS, NY, USA). A Vortex-EM silicon drift detector (Hitachi High-Technologies Science America Inc., California, USA), equipped with a collimating polycapillary optic lens (XOS, NY, USA), was placed perpendicular to the incident X-ray beam in the plane of polarization. The foci of both polycapillaries were aligned to coincide, thus obtaining a confocal detection scheme in which information can be selectively obtained from a 10(V) × 10(H) × 15(D) μm3 volume inside the sample matrix at the Fe–K edge energy. By scanning the sample through this confocal volume, direct 3D spatially resolved sample data can be collected.32–34 A schematic of the setup is shown in Fig. 1.
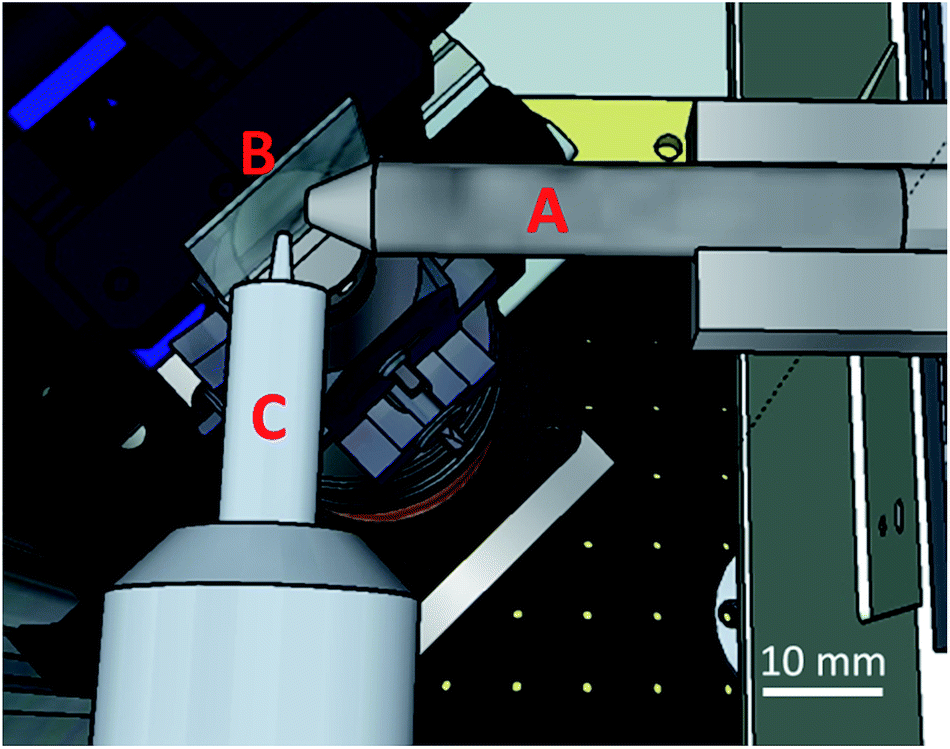 | ||
| Fig. 1 Schematic overview of the confocal micro-XAS setup at BM26A. Displayed are the focusing optic (A), focusing the primary beam on the sample (B), mounted on a goniometer head. The focus of confocal optic (C) mounted on the SDD detector coincides with that of the focusing optic, thus creating a confocal volume from which information is selectively acquired. | ||
Depth resolved XANES scans were acquired in 5 μm steps for the first 50 μm of the glass surface, and an additional XANES scan was performed at a depth of 105 μm, representative for the bulk of the glasses. The reported depths correspond to the maximal depth probed by the confocal volume, e.g. a reported depth of 20 μm corresponds to the volume 5–20 μm. A single XANES scan was split up in energy as follows: from 7012.0 eV to 7092.0 eV in 3.8 eV steps, from 7093.0 eV to 7110.0 eV in 0.9 eV steps, from 7110.5 eV to 7120.5 eV in 0.5 eV steps, from 7121.0 eV to 7132.0 eV in 0.9 eV steps and from 7133.0 eV to 7343.0 eV in 3.0 eV steps. The total integration time per energy step was adjusted for each depth and sample based on a visual inspection of the obtained XANES spectrum.
Raw XANES data were normalized for incident beam flux using the primary ionization chamber signal, pre-edge subtracted using a linear function and post-edge normalized at an energy of E0 +50 eV. Additionally, the slow variations caused by the atomic absorption profile before and after the edge were subtracted to facilitate linear combination analysis spectra comparisons.35 Due to the high absorbing nature of the glass matrix and the intrinsic properties of the confocal detection method, no self-absorption effects are expected and thus were not corrected for. To obtain data on the Fe oxidation state and coordination number (CN), the pre-edge peak, originating from the 1s → 3d electron transition, was fitted using a Gaussian function on top of an arctangent background, in line with previous research performed by Wilke et al.,21 Giuli et al.22 and others.20,23–26
EXAFS data were acquired at a depth of 25 μm and 55 μm in the glass, up to 11 Å−1 in k-space using 0.05 Å−1 steps. The data was processed using the VIPER software package36 and fitted using FEFF7 (ref. 37 and 38) calculated scatter paths. A spline was fitted using the VIPER “through the knots” routine, applying 7 knots spaced evenly over the Fourier transformed k-range from 3 to 10.5 Å−1. The first coordination shell was fitted in R-space, fitting the module and imaginary space from 0.7 to 2.0 Å (before phase shift correction). During the fitting process, an amplitude reduction factor S02 equal to 0.43 was used, as determined by fitting a hematite structure to have 6 O neighboring atoms (N) around the absorbing Fe atom. Due to the strong correlation between N and S02, the Debye–Waller-like factors were constrained between 0.0025 and 0.0035 Å2.
3. Results and discussion
XANES profiles for the Al10 glass, after IX had been performed, at various depths are displayed in Fig. 2. No clear difference is visible in general between the curves, aside from the different degrees of noise. The curves corresponding to points deeper inside the glass matrix show poorer signal to noise ratios as a result of the decreased intensity measured at these points, which in turn is due to increased absorption related to the larger X-ray path length in the sample. Similar XANES curves are found for each of the ion-exchanged boroaluminosilicate glasses (not shown). When monitoring the Fe–K XANES pre-edge peak (Fig. 2 inset), subtle differences between the curves are visible. The results of the pre-edge peak surface area and position as a function of depth are shown in Fig. 3.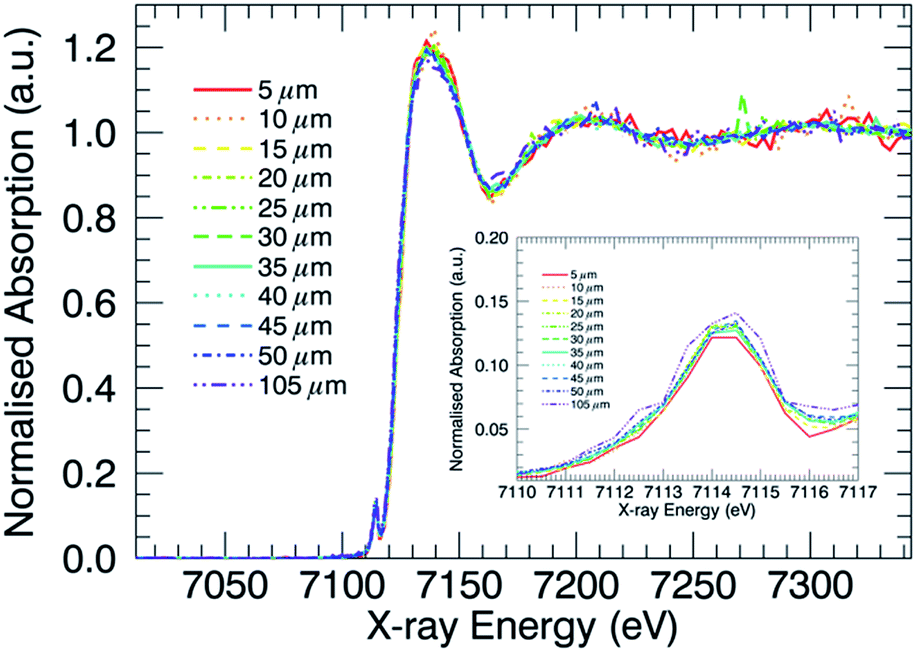 | ||
| Fig. 2 Al10 Fe–K XANES profiles as a function of probed sample depth with magnification of the pre-edge peak region in the inset, containing information on the Fe oxidation state and coordination number. | ||
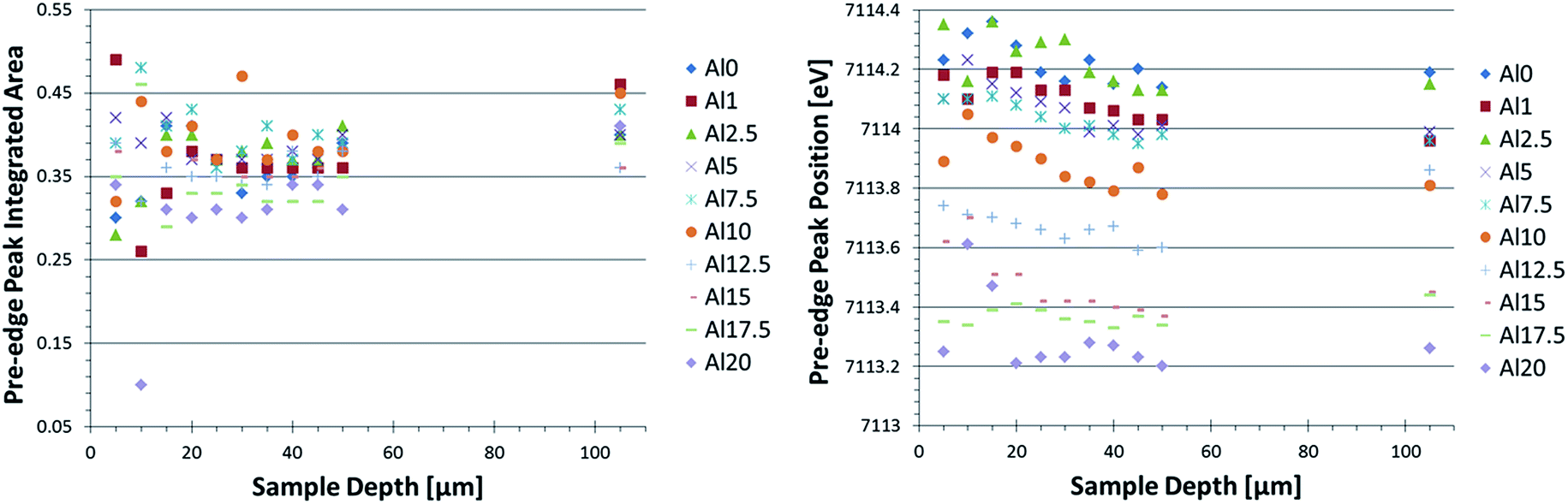 | ||
| Fig. 3 Pre-edge peak integrated area (left) and position (right) as a function of sample depth and glass composition. An increase in pre-edge peak integrated area corresponds to a decrease in coordination number; an increase in pre-edge peak position relates to an increase in oxidation state. | ||
A change in pre-edge peak position as a function of Al2O3 content is observed (Fig. 3, right image): an increase in [Al2O3]/[SiO2] ratio corresponds to a decrease in pre-edge peak position. Additionally, a slight decrease in pre-edge peak position with increasing sample depth can be seen, which is discussed in more detail below. No clear trend with pre-edge peak integrated area and glass composition or depth is observed in Fig. 3, although it appears the pre-edge peak integrated area is slightly larger in the bulk compared to the surface. This as well is discussed in greater detail below.
Since the pre-edge peak position is related to the oxidation state of Fe in the sample,20–26 a comparison of the pre-edge peak position values obtained from known structures (not displayed) shows the Fe oxidation in the glasses, which is between 3+ and 2+. A gradual decrease in oxidation state with increased [Al2O3]/[SiO2] molar concentration ratio is observed, but an exact quantification of the Fe3+/Fe2+ ratio is not performed in this case as an insufficient amount of reference compounds with known Fe3+/Fe2+ ratio were available. An attempt was made to compare using database extracted reference spectra. However, a large uncertainty in the results arose due to the difference in energy resolution and step size applied during the scans acquired from the databases.
When comparing the average pre-edge peak area and position for the surface (0–50 μm) with those in the bulk (105 μm), a more clear change in CN and oxidation state of Fe as a function of depth in the glasses is found, as shown in Fig. 4. We note that this is the average CN for both Fe2+ and Fe3+ states since it is not possible to differentiate between these two contributions using XAS. Again, the trend in oxidation state (pre-edge peak position) as a function of glass composition is clearly visible. Additionally, the bulk is generally characterized by an increased pre-edge peak area and slightly decreased pre-edge peak position, respectively, indicating a lower CN and a slightly lower oxidation state for Fe atoms in the bulk as compared to the surface.
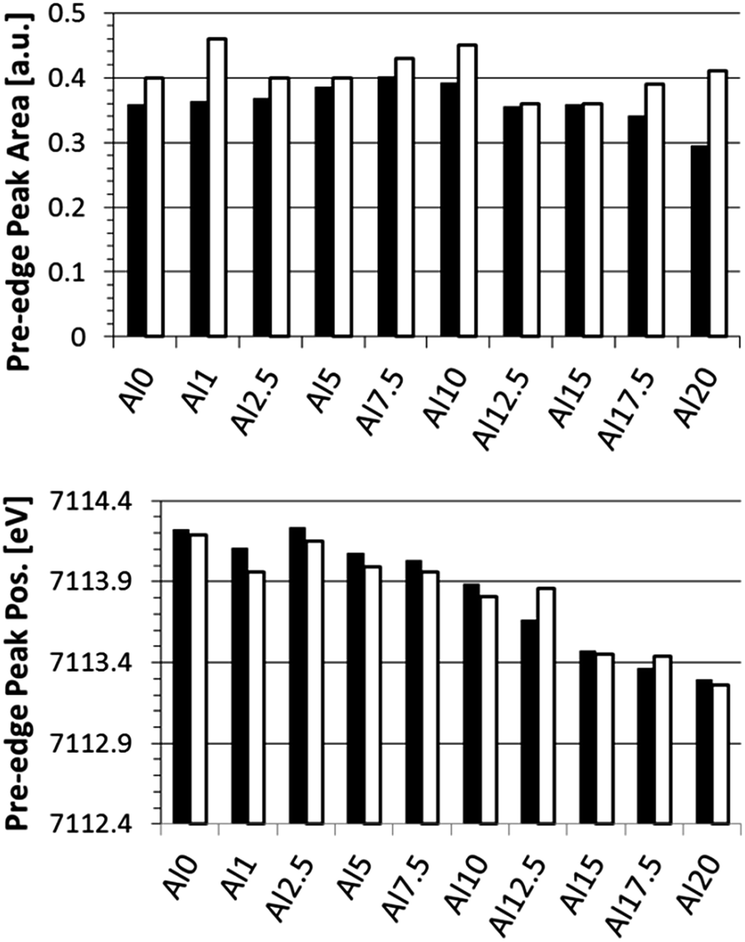 | ||
| Fig. 4 Pre-edge peak integrated area (top) and position (bottom) for the averaged surface (0–50 μm; black) and bulk (105 μm; white) regions. | ||
In order to obtain more quantitative information on the CN of Fe in the glasses, EXAFS experiments were performed in Al0, Al10, and Al20 glasses at two depths: near the surface (25 μm) and deeper in the bulk (55 μm). The data fits are shown in Fig. 5, and a graphical representation of the obtained CNs and bond distances is displayed in Fig. 6. A CN between 4 and 6 was found for Fe at the surface of the glasses, showing a fluctuation as a function of glass composition. This is in full agreement with the XANES pre-edge peak results. Generally, a higher CN is found for Fe at the surface, which is also confirmed by the XANES data (Fig. 4). Additionally, a change in Fe–O bond distance at the surface of the glasses as a function of glass composition can be seen, with shorter bonds at higher [Al2O3]/[SiO2] ratio. Deeper in the glass sample (55 μm depth), the Fe–O bond distance is found to be independent of [Al2O3]/[SiO2] ratio.
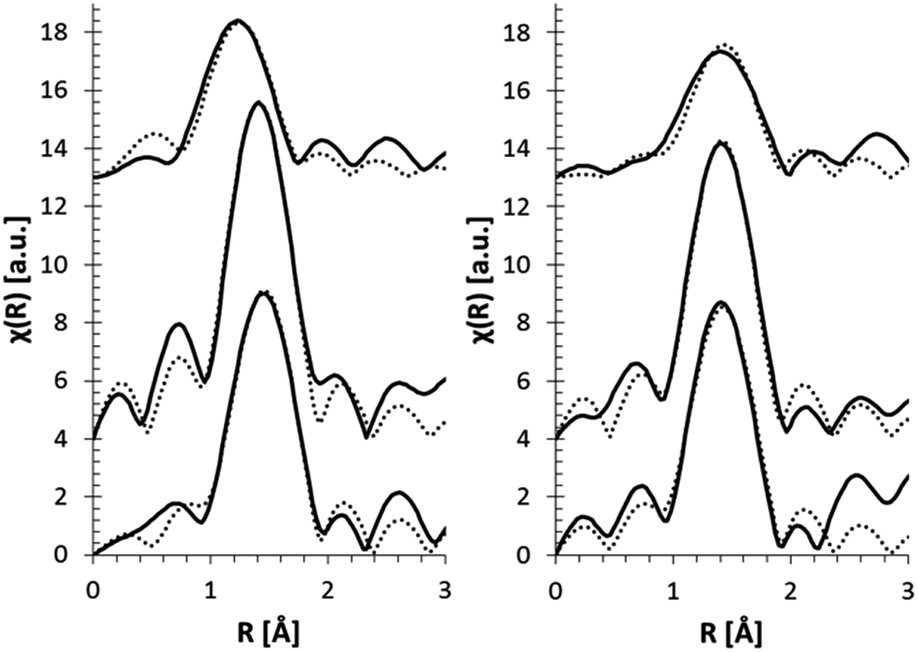 | ||
| Fig. 5 EXAFS pseudo radial distribution magnitude plots (solid curve) and their fits (dotted curve) for glasses at 25 μm (left) and 55 μm depth (right). | ||
 | ||
| Fig. 6 EXAFS fitting results for multiple glasses at two different depths. The fitted coordination number (left) and nearest neighbour Fe–O scatter path length (right) are displayed. | ||
Mysen et al. reported that an increase in Fe3+/∑Fe results in an overall decrease in CN of Fe in glasses.39 Here, a decrease in CN and oxidation state with increasing Al2O3 content was perceived, suggesting an opposite trend to what was reported by Mysen et al. It should however be noted that the glasses discussed here were ion-exchanged, resulting in a compressive stress of the surface layers. This compressive stress typically increases with [Al2O3]/[SiO2] ratio40 and may potentially lead to differences in CN for the different glasses. The change in oxidation state with glass composition on the other hand can be explained as a result of the differences in homogenization temperature for glasses with different Al2O3/SiO2 ratios, as was suggested in Smedskjaer et al.17
The decrease in CN and the increase of the Fe–O bond distances with increasing sample depth can be understood by considering the effect of the IX process on the local atomic structure. Closer to the surface more Na atoms will have been replaced by the larger K atoms, resulting in a compression of the surface layers. As the surface layers are compressed, the bond distances will decrease and even the arrangement of O atoms around Fe can change to a more tightly packed structure, increasing the Fe CN. Deeper in the sample less Na atoms will have been replaced due to the limited diffusion depth of the K atoms and a lower stress is observed, resulting in a less pronounced decrease in bond length or increase of CN. This suggests that high compressive stress plays a similar role as high pressure in the glasses.
4. Conclusions
Fe–K edge confocal micro-XANES and -EXAFS experiments have been performed to study the Fe oxidation state and coordination number (CN) as a function of depth and glass composition in a series of ion-exchanged (IX) boroaluminosilicate glasses in order to determine the structural and chemical differences of the Fe environment as a function of depth below the surface. It was found that XANES experiments are sufficient to obtain information on the oxidation state of transition metals (e.g., Fe) in these glasses, thus decreasing the exposure of the sample to radiation. In order to obtain quantitative information on the CN, EXAFS experiments are more conclusive.The Fe oxidation state decreases with increasing [Al2O3]/[SiO2] ratio in the bulk of the glasses. This feature is believed to originate from the different homogenization temperatures applied to the different glasses. Also a decrease in CN was observed with increased alumina content. Additionally, the depth-resolved data show an increase of CN and decrease of Fe–O bond distance at the surface compared to deeper inside the glass matrix, which is also explained by the IX process induced stress.
These findings represent previously unknown information on the impact of the IX-generated compressive stress and K-for-Na substitution on the local atomic structure and will aid in further development and improvement of the unique characteristics and functionalities of such damage-resistant glasses. Moreover, it is shown that confocal micro-XAS experiments are a useful addition to analytical research in order to obtain unique information on a large variation of samples, not excluding in situ or similarly challenging experiments.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Acknowledgements
We would like to thank Corning Inc. for providing us the samples to perform this study. P. Tack is funded by a Ph.D. grant of the Agency for Innovation by Science and Technology (IWT). M. Potuzak is gratefully acknowledged for suggesting this research. The Netherlands Organization for Scientific Research (NWO) and the Flanders Research Foundation (FWO) are acknowledged for providing access to the facilities of the DUBBLE beamlines at the ESRF.Notes and references
- S. R. Elliott, Physics of Amorphous Materials, Wiley, New York, 1988 Search PubMed.
- M. Guthrie, C. Tulk, C. Benmore, J. Xu, J. Yarger, D. Klug, J. Tse, H. K. Mao and R. Hemley, Phys. Rev. Lett., 2004, 93 DOI:10.1103/PhysRevLett.93.115502.
- Y. Meng, H. K. Mao, P. J. Eng, T. P. Trainor, M. Newville, M. Y. Hu, C. Kao, J. Shu, D. Hausermann and R. J. Hemley, Nat. Mater., 2004, 3, 111–114 CrossRef CAS PubMed.
- J. L. Yarger, K. H. Smith, R. A. Nieman, J. Diefenbacher, G. H. Wolf, B. T. Poe and P. F. Mcmillan, Science, 1995, 270, 1964–1967 CAS.
- M. Grimsditch, A. Polian and A. C. Wright, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 152–155 CrossRef CAS.
- S. K. Lee, P. J. Eng, H.-K. Mao, Y. Meng, M. Newville, M. Y. Hu and J. Shu, Nat. Mater., 2005, 4, 851–854 CrossRef CAS.
- L. Wondraczek, S. Sen, H. Behrens and R. E. Youngman, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76 DOI:10.1103/PhysRevB.76.014202.
- J. Wu, J. Deubener, J. F. Stebbins, L. Grygarova, H. Behrens, L. Wondraczek and Y. Yue, J. Chem. Phys., 2009, 131, 104504 CrossRef.
- V. Tyagi and A. K. Varshneya, J. Non-Cryst. Solids, 1998, 238, 186–192 CrossRef CAS.
- A. K. Varshneya, J. Non-Cryst. Solids, 1975, 19, 355–365 CrossRef CAS.
- A. Tandia, K. D. Vargheese and J. C. Mauro, J. Non-Cryst. Solids, 2012, 358, 1569–1574 CrossRef CAS.
- A. Tandia, K. D. Vargheese, J. C. Mauro and A. K. Varshneya, J. Non-Cryst. Solids, 2012, 358, 316–320 CrossRef CAS.
- K. D. Vargheese, A. Tandia and J. C. Mauro, J. Non-Cryst. Solids, 2014, 403, 107–112 CrossRef CAS.
- D. C. Boyd, US Pat., US 3778335 A, 1973.
- B. O. Myson, F. Seifert and D. Virgo, Am. Mineral., 1980, 65, 867–884 Search PubMed.
- M. M. Smedskjaer, J. C. Mauro and Y. Yue, J. Chem. Phys., 2009, 131, 244514 CrossRef PubMed.
- M. M. Smedskjaer, Q. Zheng, J. C. Mauro, M. Potuzak, S. Mørup and Y. Yue, J. Non-Cryst. Solids, 2011, 357, 3744–3750 CrossRef CAS.
- Y. Xiang, J. Du, M. M. Smedskjaer and J. C. Mauro, J. Chem. Phys., 2013, 139, 044507 CrossRef PubMed.
- Q. J. Zheng, R. E. Youngman, C. L. Hogue, J. C. Mauro, M. Potuzak, M. M. Smedskjaer and Y. Z. Yue, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86 DOI:10.1103/PhysRevB.86.054203.
- F. Farges, S. Rossano, Y. Lefrere, M. Wilke and G. E. Brown, Phys. Scr., 2005, 115, 957–959 CrossRef.
- M. Wilke, G. M. Partzsch, R. Bernhardt and D. Lattard, Chem. Geol., 2005, 220, 141–161 CrossRef CAS.
- G. Giuli, S. G. Eeckhout, E. Paris, C. Koeberl and G. Pratesi, Meteorit. Planet. Sci., 2005, 40, 1575–1580 CrossRef CAS.
- A. J. Berry, H. S. O'Neill, K. D. Jayasuriya, S. J. Campbell and G. J. Foran, Am. Mineral., 2003, 88, 967–977 CrossRef CAS.
- E. Cottrell, K. A. Kelley, A. Lanzirotti and R. A. Fischer, Chem. Geol., 2009, 268, 167–179 CrossRef CAS.
- L. Galoisy, G. Calas and M. A. Arrio, Chem. Geol., 2001, 174, 307–319 CrossRef CAS.
- W. E. Jackson, F. Farges, M. Yeager, P. A. Mabrouk, S. Rossano, G. A. Waychunas, E. I. Solomon and G. E. Brown, Geochim. Cosmochim. Acta, 2005, 69, 4315–4332 CrossRef CAS.
- S. Fuhrmann, D. Schumacher, J. Herbst and L. Wondraczek, J. Non-Cryst. Solids, 2014, 401, 82–86 CrossRef CAS.
- P. Gonçalves Ferreira, D. de Ligny, O. Lazzari, A. Jean, O. Cintora Gonzalez and D. R. Neuville, Chem. Geol., 2013, 346, 106–112 CrossRef.
- V. Martis, S. Nikitenko, S. Sen, G. Sankar, W. van Beek, Y. Filinchuk, I. Snigireva and W. Bras, Cryst. Growth Des., 2011, 11, 2858–2865 CAS.
- H. B. Stanley, D. Banerjee, L. van Breemen, J. Ciston, C. H. Liebscher, V. Martis, D. H. Merino, A. Longo, P. Pattison, G. W. M. Peters, G. Portale, S. Sen and W. Bras, CrystEngComm, 2014, 16, 9331–9339 RSC.
- S. Nikitenko, A. M. Beale, A. M. van der Eerden, S. D. Jacques, O. Leynaud, M. G. O'Brien, D. Detollenaere, R. Kaptein, B. M. Weckhuysen and W. Bras, J. Synchrotron Radiat., 2008, 15, 632–640 CrossRef CAS PubMed.
- G. Silversmit, B. Vekemans, S. Nikitenko, W. Bras, V. Czhech, G. Zaray, I. Szaloki and L. Vincze, J. Synchrotron Radiat., 2009, 16, 237–246 CrossRef CAS PubMed.
- G. Silversmit, B. Vekemans, S. Nikitenko, S. Schmitz, T. Schoonjans, F. E. Brenker and L. Vincze, Phys. Chem. Chem. Phys., 2010, 12, 5653–5659 RSC.
- G. Silversmit, B. Vekemans, S. Nikitenko, K. Tirez, W. Bras, F. E. Brenker and L. Vincze, presented in part at the 14th International Conference on X-Ray Absorption Fine Structure (XAFS14), 2009 Search PubMed.
- S. Calvin, XAFS for Everyone, Taylor & Francis, 2013 Search PubMed.
- K. V. Klementev, J. Phys. D: Appl. Phys., 2001, 34, 2241–2247 CrossRef CAS.
- A. L. Ankudinov, PhD thesis, University of Washington, 1996.
- A. L. Ankudinov and J. J. Rehr, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, R1712–R1715 CrossRef CAS.
- B. O. Mysen, Geochim. Cosmochim. Acta, 2006, 70, 2337–2353 CrossRef CAS.
- M. J. Dejneka, J. C. Mauro, M. Potuzak, M. M. Smedskjaer, and R. E. Youngman, US Pat., US 9,145,333 B1, 2015.
| This journal is © The Royal Society of Chemistry 2016 |