
Polymeric micelles from block copolymers containing 2,6-diacylaminopyridine units for encapsulation of hydrophobic drugs†
Alberto Concellóna,
Rafael Clavería-Gimenobc,
Adrián Velázquez-Campoycde,
Olga Abianbcdf,
Milagros Piñol*a and
Luis Oriol*a
aDepartamento de Química Orgánica, Facultad de Ciencias, Instituto de Ciencia de Materiales de Aragón (ICMA), Universidad de Zaragoza-CSIC, 50009, Zaragoza, Spain. E-mail: mpinol@unizar.es; loriol@unizar.es
bInstituto Aragonés de Ciencias de la Salud (IACS), Zaragoza, Spain
cIIS Aragón, Zaragoza, Spain
dInstitute of Biocomputation and Physics of Complex Systems (BIFI), Joint Unit IQFR-CSIC-BIFI, Universidad de Zaragoza, Zaragoza, Spain
eFundación ARAID, Government of Aragón, Spain
fCentro de Investigación Biomédica en Red en el Área Temática de Enfermedades Hepáticas y Digestivas (CIBERehd), Spain
First published on 24th February 2016
Abstract
The paper describes the synthesis and characterization of novel amphiphilic block copolymers composed of a hydrophilic (polyethylene glycol) block and a hydrophobic polymetacrylate block with pendant 2,6-acylaminopyridine units. As demonstrated by TEM, these polymers are capable of forming stable spherical micelles in water with diameters of 18–25 nm, depending on the average molar mass, determined by dynamic light scattering. The polymeric micelles are sensitive to pH and can encapsulate hydrophobic molecules such as Nile Red. The cytotoxicity of the block copolymers has been tested in HeLa and Lunet cell lines and proved to be adequate. The polymeric micelles have been used to load camptothecin (CPT), an effective antiviral against hepatitis C virus (HCV), by simple physical diffusion. The antiviral activity against HVC of these CPT-loaded polymeric micelles has been tested on Huh 5-2 Lunet cells, demonstrating the potential of these polymers as nanocarriers for drug delivery applications.
Introduction
Polymeric drug carriers are one of the current challenges of nanomedicine. Since the concept of physical drug encapsulation within polymeric aggregates was introduced, a significant number of polymer assemblies have been identified. In particular, the construction of amphiphilic block copolymer-based drug carriers is a subject of great interest and a stimulating topic of interdisciplinary research in chemistry, biology and materials science. In aqueous media, self-assembly of amphiphilic block copolymers (BCs) to minimize energetically unfavorable hydrophobic water interactions can lead to a variety of polymeric nanostructures including especially appealing spherical micelles and vesicles.1–7Spherical micelles, with average diameters of 5–100 nm, typically exhibit core–shell architectures in which a hydrophilic shell affords water solubility and a hydrophobic core provides an ideal location for encapsulation of hydrophobic molecules.8–12 As far as intravenous delivery of chemotherapeutic agents is concerned, polymeric micelles are perfect instruments to solubilize hydrophobic drugs, infiltrate the body with a minimal immunogenic response and target diseased tissues, thus releasing the drug payload at its destination while minimizing damage to healthy tissues.13,14 Indeed, in polymeric micelles, encapsulation is dictated by the chemical and physical nature of the core, which in turn depends on the nature of the core forming hydrophobic block and of the drug. However there are additional factors such as the stability of micelles, amongst other, that might limit their application as drug delivery carriers.15–17 These issues can be handled by the introduction of non-covalent interactions inside the core of the polymeric micelle to strengthen micelles or even endow them with certain functionalities.18 Amongst different alternatives, hydrogen bonding that is a high specific and directional supramolecular interaction might be used to direct the self-assembly of BCs and stabilize the micellar cores. With this idea, amphiphilic BCs possessing bases of nucleic acids as side groups, which form reinforced polymeric micelles through hydrogen bonding, have been reported.19–21 Also, urea-based polymers forming stable micelles enhanced by the presence of hydrogen bonds between urea groups have been prepared. Besides, drugs have been more efficiently loaded into these micelles, by linking the drug to the urea groups through hydrogen bonding.22–24
In this work, we describe the synthesis and thermal properties of new amphiphilic BCs that have a polymethacrylate block bearing 2,6-diacylaminopyridine pendant units (DAP) and a poly(ethylene glycol) block (PEG) of two different molar masses, 2000 and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (PEG2-b-PDAP and PEG10-b-PDAP, respectively). The presence in the hydrophobic block of 2,6-diacylaminopyridine, a nucleobase analogue capable of forming multiple hydrogen bonds, may play an important role in micelles reinforcement and simultaneously providing the ability to respond to the pH. We have investigated the morphology of the aggregates formed in water and their pH-dependent behavior. Additionally, the potential of these BCs for effective loading and delivering has been tested using camptothecin (CPT), a hydrophobic drug. It has been demonstrated that non cytotoxic CPT-loaded polymer micelles display similar anti-HCV activity than free, and more toxic, CPT.
000 (PEG2-b-PDAP and PEG10-b-PDAP, respectively). The presence in the hydrophobic block of 2,6-diacylaminopyridine, a nucleobase analogue capable of forming multiple hydrogen bonds, may play an important role in micelles reinforcement and simultaneously providing the ability to respond to the pH. We have investigated the morphology of the aggregates formed in water and their pH-dependent behavior. Additionally, the potential of these BCs for effective loading and delivering has been tested using camptothecin (CPT), a hydrophobic drug. It has been demonstrated that non cytotoxic CPT-loaded polymer micelles display similar anti-HCV activity than free, and more toxic, CPT.
Experimental
Materials and characterization
Monomer DAP was synthesized according to previously reported procedures.25 All other commercially reagents were purchased from Sigma-Aldrich and used as received. Poly(ethylene glycol) 4-cyano-4-(phenylcarbonothioylthio) pentanoate reversible addition–fragmentation chain transfer agents of 2000 g mol−1 (PEG2-CTA) or 10200 g mol−1 (PEG10-CTA) molar masses were purchased from Sigma-Aldrich, the average molar masses were verified by MALDI-TOF and 1H NMR.
FTIR spectra were obtained on a Bruker Vertex 70 FT-IR spectrophotometer using KBr pellets, in the 4000–400 cm−1 region, with 4 cm−1 accuracy. Solution NMR experiments were carried out on Bruker Avance spectrometers operating at 400 MHz for 1H, using standard pulse sequences. Chemical shifts are given in ppm relative to TMS and the solvent residual peak was used as internal reference. Elemental analysis was performed using a Perkin-Elmer 2400 microanalyzer. MALDI-TOF MS was performed on an Autoflex mass spectrometer (Bruker Daltonics) using dithranol as matrix. Size exclusion chromatography (SEC) was carried out on a Waters e2695 Alliance liquid chromatography system equipped with a Waters 2424 evaporative light scattering detector using two Styragel® columns, HR4 and HR1 from Waters. Measurements were performed in THF using a flow of 1 mL min−1 and poly(methyl methacrylate) (PMMA) narrow molar mass standards. Thermogravimetric analysis (TGA) were performed using a Q5000IR from TA instruments at heating rate of 10 °C min−1 under a nitrogen atmosphere. Thermal transitions were determined by differential scanning calorimetry (DSC) using a DSC Q2000 from TA instruments with powdered samples (2–5 mg) sealed in aluminum pans. Glass transition temperatures (Tg) were determined at the half height of the baseline jump, and first order transition temperatures were read at the maximum of the corresponding peak. UV-vis absorption spectra were measured with a UV4-200 from ATI-Unicam. Fluorescence measurements were performed using a Perkin Elmer LS 50B fluorescence spectrophotometer.
General procedure for the synthesis of PEGx-b-PDAP polymers
Monomer DAP (0.30 g, 0.80 mmol), poly(ethylene glycol) 4-cyano-4-(phenylcarbonothioylthio) pentanoate (PEGx-CTA) (for x = 2: 0.16 g, 0.08 mmol; for x = 10: 0.16 g, 0.02 mmol), 2,2′-azobisisobutyronitrile (AIBN) (for x = 2: 2.0 mg, 0.012 mmol; for x = 10: 0.39 mg, 0.002 mmol) and N,N-dimethylformamide (DMF) (1.25 mL) were added to a Schlenk flask that was then closed with a rubber septum. The flask was deoxygenated by three freeze–pump–thaw cycles and flushed with argon. The reaction mixture was stirred at 80 °C. After 2.5 h (for x = 2) or 5 h (for x = 10), the mixture was quenched with liquid nitrogen and then carefully precipitated using cold diethyl ether. The polymer was dried in vacuum at room temperature for 24 h.Characterization data of PEG2-b-PDAP
Yield: 55%. IR (KBr) ν (cm−1): 3321, 1736, 1588, 1522, 1449, 1243, 1151. 1H NMR (400 MHz, CDCl3) δ (ppm): 8.89–8.30 (m, 2H), 7.94–7.54 (m, 3H), 4.38–4.04 (m, 4H), 3.87–3.41 (m, 16H), 2.85–2.58 (m, 4H), 2.40–2.24 (m, 2H), 2.02–1.76 (m, 2H), 1.50–0.75 (m, 6H). Anal. calcd for C284H425N31S2O107: C, 56.31%; H, 7.07%; N, 7.17%; S, 1.06%. Found: C, 56.04%; H, 7.40%; N, 7.14%; S, 1.03%.Characterization data of PEG10-b-PDAP
Yield: 76%. IR (KBr) ν (cm−1): 3320, 1736, 1587, 1523, 1449, 1244, 1151. 1H NMR (400 MHz, CDCl3) δ (ppm): 8.97–8.40 (m, 2H), 7.93–7.50 (m, 3H), 4.41–4.00 (m, 4H), 3.76–3.54 (m, 18H), 2.93–2.59 (m, 4H), 2.39–2.17 (m, 2H), 2.02–1.76 (m, 2H), 1.35–0.69 (m, 6H). Anal. calcd for C1296H1980N139S2O505: C, 56.27%; H, 7.22%; N, 7.04%; S, 0.23%. Found: C, 56.23%; H, 7.57%; N, 7.07%; S, 0.56%.Preparation of the self-assemblies in water and characterization
Milli-Q water was gradually added to a solution of 5 mg mL−1 of PEGx-b-PDAP in THF. The process was followed by measuring the turbidity and when a constant value of turbidity was reached, the suspension was dialyzed against water to remove the organic solvent using a Spectra/Por® dialysis membrane (MWCO 1000) for 4 days. Aqueous suspensions of the micelles with a concentration around 1.25 mg mL−1 (PEG2-b-PDAP) or 1.15 mg mL−1 (PEG10-b-PDAP) were obtained. The morphological study of the block copolymer micelles was carried out by transmission electron microscopy (TEM) using a JEOL-2000 FXIII and TECNAI G2 20° (FEI COMPANY) electron microscope operating at 200 kV. Preparation of samples for TEM inspection: 5 μL of a 0.75 mg mL−1 water micelles dispersion was deposited onto carbon-coated copper grid. Water of the sample was removed by capillarity using filter paper. Then, the sample was stained with uranyl acetate and the grid was left to dry overnight under vacuum. Dynamic light scattering (DLS) measurements were carried out on a Malvern Instrument Nano ZS using a He–Ne laser with a 633 nm wavelength, a detector angle of 173° at 25 °C using a He–Ne laser with a 633 nm wavelength. The self-assemblies concentrations were 0.075 mg mL−1 and size measurements were performed at least three times on each sample to ensure consistency.Determination of the critical aggregation concentration (CAC)
Critical aggregation concentration (CAC) was determined by fluorescence spectroscopy using Nile Red following a previously described method.26 119 μL of a solution of Nile Red in DCM (5 × 10−6 M) was added into a series of flasks and the solvent was evaporated. Afterwards, a water micellar suspension with concentration ranging from 1.0 × 10−4 to 1.0 mg mL−1 was added to each flask. The micelles suspensions were prepared by diluting the former 1.25 mg mL−1 (PEG2-b-PDAP) or 1.15 mg mL−1 (PEG10-b-PDAP) suspension. In each flask a final concentration of 10−6 M of Nile Red was reached. These solutions were stirred overnight to reach equilibrium before fluorescence was measured. The emission spectra of Nile Red were registered from 560 to 700 nm while exciting at 550 nm.Evaluation of the pH effect on the micelles stability
PEG2-b-PDAP or PEG10-b-PDAP micellar suspensions (1 mL, 1.25 mg mL−1 for PEG2-b-PDAP or 1.15 mg mL−1 for PEG10-b-PDAP) were dispersed in a sodium acetate/acetic acid (pH = 5.5, 0.05 M) or citric acid/sodium citrate (pH = 3.0, 0.05 M) buffer solutions. The suspensions were stirred for 5 min at room temperature. Micellar suspensions with a polymer concentration of 0.75 mg mL−1 were obtained.Encapsulation of camptothecin (CPT)
A suspension of the micelles (1 mL, 1.25 mg mL−1 for PEG2-b-PDAP or 1.15 mg mL−1 for PEG10-b-PDAP) and CPT in DMSO (1 mL, 1.0 mg mL−1) were stirred overnight to reach equilibrium before dialysis against water using a Spectra/Por® dialysis membrane (MWCO 1000) for five days at room temperature to replace DMSO. After dialysis the mixture was filtered using a Corning® syringe filters (surfactant-free cellulose acetate membrane, membrane diam. 28 mm, pore size 0.45 μm) to remove precipitated CPT. Under these conditions, a water suspension of CPT charged micelles with a polymer concentration of around 0.56 mg mL−1 for PEG2-b-PDAP or 0.58 mg mL−1 for PEG10-b-PDAP were obtained. The CPT loading content of these micelles was determined as follows: an aliquot of CPT-loaded micelles (20 μL) was lyophilized, the resulting product was dissolved in a 2 mL volumetric flask using DMSO. The amount of CPT was determined by emission spectroscopy (λexc = 365 nm, λem = 438 nm), using a calibration curve previously obtained.The percentage of drug loading content (DLC) was calculated according to the following equation:

Isothermal titration calorimetry (ITC) assay
The process of CPT loading into PEG2-b-PDAP and PEG10-b-PDAP polymeric micelles was studied by ITC using with a high-sensitivity isothermal titration VP-ITC microcalorimeter (MicroCal, USA). Experiments were performed at 25 °C in aqueous media. A 100 μM solution of CPT placed in the calorimetric cell was titrated with sequential injections of a 30 μM PEG2-b-PDAP or PEG10-b-PDAP micellar suspension. Control experiments were performed under the same experimental conditions. The heat evolved after each injection was obtained from the integral of the calorimetric signal. The heat due to the binding reaction was obtained as the difference between the reaction heat and the corresponding heat of dilution, the latter estimated as a constant heat throughout the experiment, and included as an adjustable parameter in the analysis. The association constant (Ka) and the enthalpy change (ΔH) were obtained through non-linear regression of experimental data to a model considering one class of ligand binding sites. Data were analyzed using software developed in our laboratory implemented in Origin 7 (OriginLab, USA).Cells and replicon system
The highly permissive cell clone HeLa, Huh 7-Lunet, as well as Huh 7 cells containing subgenomic HCV replicons I389luc-ubi-neo/NS3-3′/5.1 (Huh 5-2), I377NS3-3′/wt (Huh 9-13) or I389/hygro-ubi-NS3-3/5.1 (a kind gift from Dr V. Lohmann and Dr R. Bartenschlager) have been used.27,28 Cells were grown in Dulbecco's modified Eagle's medium (DMEM; Gibco, Belgium) supplemented with 10% heat-inactivated fetal bovine serum (PAN-Biotech GmbH, Germany), 1× non-essential amino acids (Gibco), 100 IU mL−1 penicillin (Gibco), 100 μg mL−1 streptomycin (Gibco), and 250 μg mL−1 geneticin (G418; Gibco).Antiviral assays with Huh 5-2 cells
Antiviral assays for assessing the efficacy of the target compounds were performed as previously described.29–31 Briefly, Huh 5-2 cells were seeded at a density of 7 × 103 cells per well in a tissue culture-treated white 96-well view plate (Techno Plastic Products AG, Switzerland) in complete DMEM supplemented with 250 μg mL−1 G418. After incubation for 24 h at 37 °C, medium was removed and 2-fold serial dilutions up to 1 μM in complete DMEM of the PEG2-b-PDAP-CPT and PEG10-b-PDAP-CPT complexes were added in a total volume of 100 μL. After 3 days of incubation at 37 °C, cell culture medium was removed and luciferase activity was determined using the Bright-GloTM Luciferase Assay System (Promega Corporation, The Netherlands). The luciferase signal was measured using a Synergy HT Multimode Reader (BioTek Instruments Inc, USA). The 50% effective concentration (EC50) was defined as the concentration of compound that reduced the luciferase signal by 50%.Cytostatic assays
Cytostatic assays for assessing the cell viability of the PEG2-b-PDAP-CPT and PEG10-b-PDAP-CPT complexes were performed as previously described.27,28,30 Briefly, Huh 7-Lunet, HeLa and Huh 5-2 cell lines were seeded at a density of 7 × 103 cells per well of a 96-well plate in complete DMEM (with the appropriate concentrations of G418, in case of Huh 5-2). Serial dilutions of the test compounds up to 1 μM in complete DMEM were added 24 hours after seeding. Cells were allowed to proliferate for 3 days at 37 °C, after which the cell number was determined by CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega Corporation). The 50% cytostatic concentration (CC50) was defined as the concentration that inhibited the proliferation of exponentially growing cells by 50%.Results and discussion
Synthesis and characterization of amphiphilic BCs
Two diblock copolymers (PEG2-b-PDAP and PEG10-b-PDAP) were prepared by reversible addition–fragmentation chain transfer (RAFT) polymerization of the methacrylic monomer DAP using commercially available poly(ethylene glycol) 4-cyano-4-(phenylcarbonothioylthio)pentanoate macro-CTAs (Fig. 1) with average number molar masses (Mn) of approx. 2000 g mol−1 (PEG2-CTA) or 10200 g mol−1 (PEG10-CTA). Mn of these macro-CTAs were determined by MALDI-TOF and were in agreement with those reported by the supplier. Monomer DAP was easily synthesized in two steps from commercially available compounds.25 The polymerizations were adjusted to obtain hydrophilic/hydrophobic weight ratios of around 35/65 (Table 1), to favor the formation of spherical micelles of amphiphilic BCs in water.4,5 Average number molar masses of the BCs were determined by 1H NMR spectroscopy (MNMRn in Table 1 and Fig. S1†). The polymerizations were well controlled as demonstrated by the close agreement between experimental molar masses (MNMRn) and those calculated by considering a monomer conversion of 1 (Mtheon in Table 1). The relative average number molar masses of PEGx-b-PDAP were also estimated by SEC using PMMA standards (MSECn in Table 1). Dispersities (Đ), also determined by SEC, were in agreement with data expected for a controlled radical polymerization (Đ < 1.1).
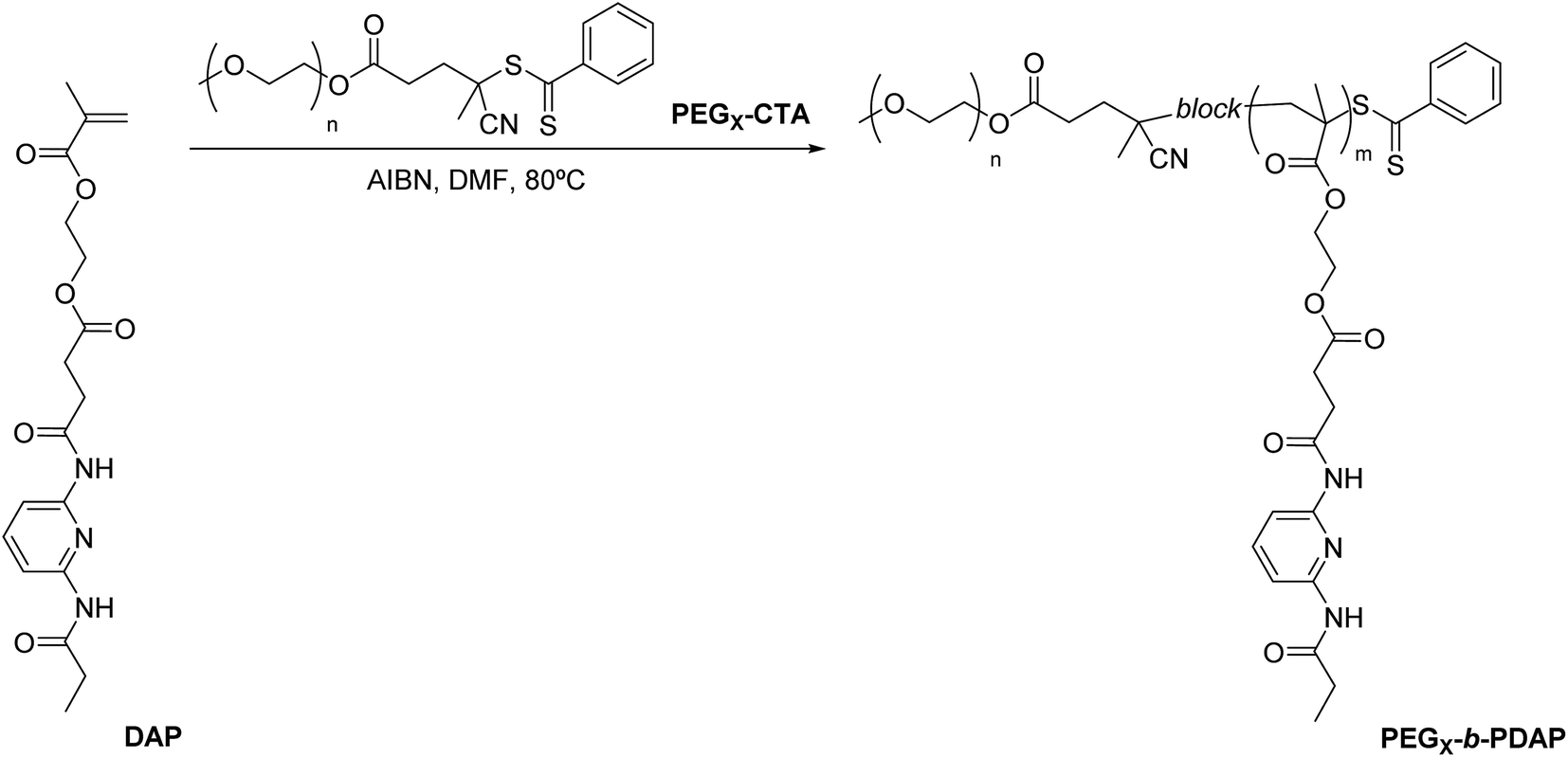 | ||
| Fig. 1 Synthesis of PEGx-b-PDAP block copolymers. | ||
| Polymer | Philic/phobic ratioa | MMSnb (g mol−1) | MNMRnc (g mol−1) | [M]/[CTA]d | Mtheone (g mol−1) | MSECnf (g mol−1) | Đf |
|---|---|---|---|---|---|---|---|
| a Hydrophobic/hydrophilic ratio in BCs is given in weight percentage considering the PEG block as hydrophilic and the PDAP block as hydrophobic.b Number-average molar mass (Mn) of commercial PEG (Sigma-Aldrich) calculated from MALDI-TOF mass spectrometry.c Mn calculated as the sum of molar masses of the PDAP block (determined by NMR) and the PEG block (determined by MALDI-TOF mass spectrometry, MMSn).d Feed ratio of monomer to PEGx-CTA macroinitiator.e Calculated according to formula: Mtheon = ([M]/[CTA] × conv × Mmonomer) + MCTA.f Relative Mn and Đ calculated by SEC using THF (1 mL min−1) and PMMA standards. | |||||||
| PEG2-CTA | — | 2000 | — | — | — | 3100 | 1.02 |
| PEG2-b-PDAP | 35/65 | — | 5800 | 10 | 5700 | 6800 | 1.08 |
| PEG10-CTA | — | 10200 |
— | — | — | 14300 |
1.04 |
| PEG10-b-PDAP | 37/63 | — | 27500 |
50 | 29000 |
20700 |
1.05 |
The thermal stability was studied by thermogravimetric analysis (TGA) using powdered samples. All the samples showed good thermal stability with onset temperatures (Tonset) associated to mass loss above 200 °C (Table 2). Evolution of volatiles due to the presence of residual solvents or water was not observed. The thermal transitions were evaluated by differential scanning calorimetry (DSC) and the relevant parameters are gathered in Table 2. The PEG macro-CTAs that are the precursors of the PEG block in the BCs, PEGx-CTA, showed reversing melting/crystallization processes. The homopolymer PDAP, which was previously synthesized, was an amorphous material with Tg at 73 °C.25 However, instead of the expected transitions associated to the two constitutive blocks, both PEGx-b-PDAP BCs exhibited only a glass transition below room temperature, which indicates miscibility between these polymeric blocks. This behavior was also observed in the corresponding physical blends of PDAP homopolymer and the PEGx-CTAs with the same mass fraction as in PEGx-b-PDAP BCs, and prepared by solvent evaporation from THF (Fig. 2) as reference. Thus, this confirms the, at least partial, miscibility of the PEG and PDAP segments in bulk.
| Polymer | Tonseta (°C) | Tgb (°C) | Tmc (°C) |
|---|---|---|---|
| a Onset temperature associated to mass loss detected in the thermogravimetric curve.b Glass transition temperature determined at the half height of the baseline jump on the second heating scan at 10 °C min−1.c Melting temperature read at the maximum of the peak on the second heating scan at 10 °C min−1.d Physical blend of PEGx-CTA and PDAP homopolymer. | |||
| PDAP | 255 | 73 | — |
| PEG2-CTA | 210 | — | 48 |
| PEG2-b-PDAP | 250 | 6 | — |
| PEG2-CTA + PDAPd | 260 | 0 | — |
| PEG10-CTA | 310 | — | 58 |
| PEG10-b-PDAP | 255 | −3 | — |
| PEG10-CTA + PDAPd | 250 | −6 | — |
 | ||
| Fig. 2 DSC traces corresponding to the second heating scan of (a) PEG2-CTA, (b) PDAP homopolymer, (c) blend of PEG2-CTA and PDAP and (d) PEG2-b-PDAP (10 °C min−1, Exo down). | ||
Self-assembly of amphiphilic BCs in water
Self-assembled structures of PEG2-b-PDAP and PEG10-b-PDAP were prepared by the co-solvent method using THF/water and following the micellization process by turbidimetry. Amphiphilic BCs were first dissolved in THF (a good solvent for both blocks) and then water, which is non solvent for the hydrophobic block, was slowly added. At some point of the addition a sudden jump in turbidity occurred indicating that polymer self-assembly starts. Once turbidity reached an almost constant value, the resulting dispersion was dialyzed against water to remove the organic solvent. After dialysis, stable micellar suspensions were obtained.The morphology of the PEG2-b-PDAP and PEG10-b-PDAP self-assemblies was first investigated by transmission electron microscopy (TEM). TEM images (Fig. 3a) evidence the presence of spherical micelles with diameter of approx. 18 nm for PEG2-b-PDAP, and 25 nm for PEG10-b-PDAP. The average size of the micelles was also determined by dynamic light scattering (DLS) and mean hydrodynamic diameters (Dh) of 21 nm and 32 nm were found for PEG2-b-PDAP and PEG10-b-PDAP, respectively (Fig. 4).
 | ||
| Fig. 3 TEM images of PEG2-b-PDAP (left) and PEG10-b-PDAP (right): (a) initial micelles, (b) micelles at pH = 5.5, (c) micelles at pH = 3 and (d) CPT-loaded micelles. Scale bar = 100 nm. | ||
 | ||
| Fig. 4 DLS measurements of a water suspension of: (a) PEG2-b-PDAP and (b) PEG10-b-PDAP micelles, initially measured and after loading with camptothecin. | ||
The critical aggregation concentration (CAC) in water of PEG2-b-PDAP and PEG10-b-PDAP was determined by fluorescence spectroscopy using Nile Red as a polarity sensitive probe (Fig. S2†).26 Micellar suspensions of amphiphilic BCs were stirred overnight together with Nile Red at room temperature and the emission spectra of Nile Red were registered from 560 to 700 nm. Calculated CAC values of PEG2-b-PDAP and PEG10-b-PDAP were 27 and 25 μg mL−1, respectively, which are typical for amphiphilic BCs.32
The core of the micelles is formed by the hydrophobic PDAD block which might confer sensitivity to the pH in a way that micelles might swell or even dissociate at acidic pH values.33 To evaluate the stability of the micelles against acidic pH, PEG2-b-PDAP and PEG10-b-PDAP micellar suspensions were dispersed into pH = 5.5 and pH = 3.0 buffer solutions. TEM images show that at pH = 5.5, there is a partial disturbance of the micellar self-assemblies for PEG2-b-PDAP, while PEG10-b-PDAP micelles are almost unaltered (Fig. 3b). However, at pH = 3.0 (Fig. 3c) protonation of the nitrogen atom of the pyridine ring in the 2,6-diacylaminopyridine unit increases the repulsive forces between the hydrophobic segments, resulting in the micelles disruption for both polymers. Due to its hydrophobic nature, Nile Red is retained into the core of the micelle, as demonstrated in the CAC experiments. Thus, micellar suspensions, having concentrations above the CAC, were stirred with Nile Red (10−6 M) in buffered media. The emission recorded at 560–700 nm (exciting at 550 nm) for Nile Red loaded micelles (Fig. 5) strongly decreases for PEG2-b-PDAP in particular at pH = 3.0, indicating that the local environment of Nile Red is mainly hydrophilic and, therefore, corroborating distortion of the micelles. For PEG10-b-PDAP the emission decrease is only moderated at pH = 5.5 confirming again TEM observations.
 | ||
| Fig. 5 Emission spectra of Nile Red encapsulated micelles of (a) PEG2-b-PDAP and (b) PEG10-b-PDAP. | ||
Encapsulation of camptothecin
The potential ability of PEG2-b-PDAP and PEG10-b-PDAP micelles to load and deliver hydrophobic drugs was tested using CPT. Micelles were loaded with CPT by simple physical diffusion when stirring the polymeric micellar suspension with a CPT solution. Aqueous suspensions of CPT-loaded micelles (PEG2-b-PDAP-CPT and PEG10-b-PDAP-CPT) were thus obtained. The UV-vis spectra of aqueous suspensions of unloaded and CPT-loaded micelles are compared in Fig. 6a. The intense band around 295 nm was assigned to the π–π* transition of the pyridine unit. Additionally, significant absorption peaks at 253 and 365 nm were observable in the spectrum of the CPT-loaded micelles due to entrapped CPT. The emission spectra of CPT-loaded micelles showed a band at 438 nm (λexc = 365 nm), while no evidence of emission was detected for unloaded micelles (Fig. 6b). Drug loaded contents on these micellar aqueous dispersions were determined by emission spectroscopy giving values of 1.79 μg mL−1 (corresponding to 3.204 μg of CPT per mg of BC, DLC = 0.32%) and 2.51 μg mL−1 (4.320 μg of CPT per mg of BC, DLC = 0.43%) for PEG2-b-PDAP-CPT and PEG10-b-PDAP-CPT, respectively. | ||
| Fig. 6 (a) UV-vis spectra and (b) fluorescence spectra (λexc = 365 nm) of PEG2-b-PDAP. | ||
The CPT encapsulation process was studied by isothermal titration calorimetry (ITC). Experiments were recorded at 25 °C while a 100 μM CPT solution was titrated with sequentially programmed injections of a 30 μM micellar suspensions. The association constant (Ka) and the thermodynamic parameters for the interaction of PEG2-b-PDAP and PEG10-b-PDAP micelles with CPT were evaluated from the registered calorimetric signal (Table 3). Results showed that the interaction process is energetically favorable for both PEG2-b-PDAP and PEG10-b-PDAP with a Gibbs energy of binding (ΔG) of −10 kcal mol−1 and similar affinity constants (Ka) 1.5–1.8 × 107. The interaction was driven by the entropic contribution (ΔS), which it is related to unspecific, likely hydrophobic, interactions between the CPT and the internal hydrophobic core of the polymeric micelles. This is the most common situation described for poorly water soluble drugs.34
| Polymer | Ka | ΔG (kcal mol−1) | ΔH (kcal mol−1) | TΔS (kcal mol−1) |
|---|---|---|---|---|
| a Constant affinity parameter (Ka) and thermodynamic data of the process determined by ITC at 25 °C in aqueous media. | ||||
| PEG2-b-PDAP | 1.5 × 107 ± 6.9 × 106 | −9.8 ± 0.1 | 7.0 ± 0.5 | 17.0 ± 0.5 |
| PEG10-b-PDAP | 1.8 × 107 ± 7.0 × 106 | −9.9 ± 0.1 | 9.0 ± 0.5 | 19.0 ± 0.5 |
The morphology of the CPT-loaded micelles was investigated by TEM. Fig. 3d shows spherical micelles with a diameter of approx. 25 nm (PEG2-b-PDAP-CPT) and 36 nm (PEG10-b-PDAP-CPT). The average size was also evaluated by DLS and Dh values of 30 nm and 42 nm were found for CPT-loaded PEG2-b-PDAP and PEG10-b-PDAP micelles, respectively (Fig. 4). It is noteworthy that the encapsulated hydrophobic drug provokes an increase in the size of the micelles when compared to unloaded micelles of approx. 67–68% without changing their morphology or their stability.
Toxicity and antiviral activity of CPT-loaded polymeric BCs in cell culture
Previous to any further experiment, the cytotoxicity of the amphiphilic BCs was tested in cell culture using two different cell lines, Lunet and HeLa. Results for PEG2-b-PDAP are represented in Fig. 7 but there was not any remarkable difference in behavior between PEG2-b-PDAP and PEG10-b-PDAP (see Fig. S3 in the ESI†). In the case of Lunet cells, there was not a decrease in cell viability after 72 h of cell culture in presence of increasing concentrations of PEG2-b-PDAP (from 0.016 to 1 mg mL−1). For HeLa, a different cell line, there was a slight effect when cells were incubated for 72 h using polymer concentrations above 0.25 mg mL−1. At 1 mg mL−1 and 72 h of cell culture, the viability was 30% lower respect to the controls. Accordingly, HeLa cell line seems to be slightly more sensitive to these polymers than the hepatic cell line (Lunet).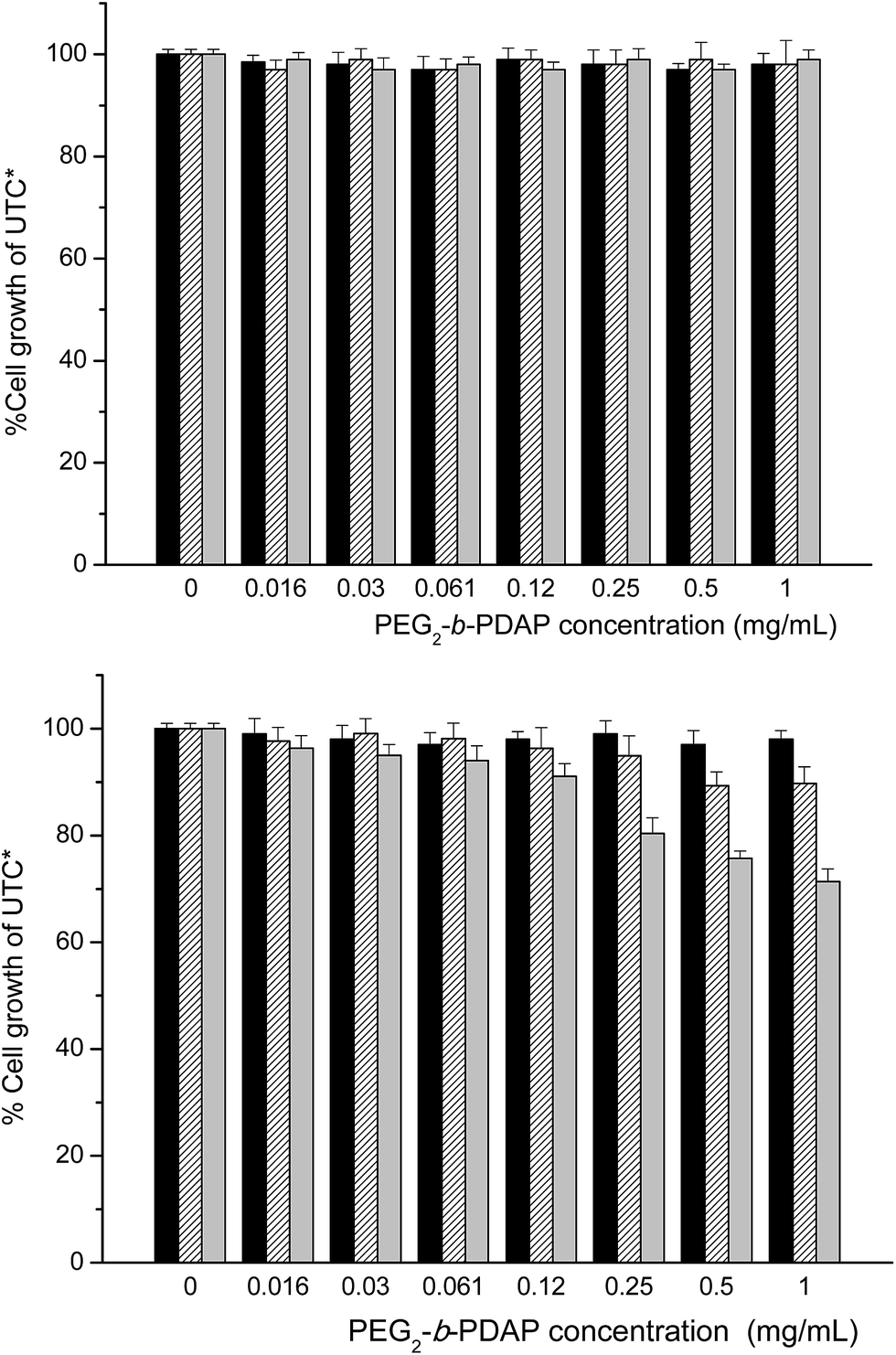 | ||
| Fig. 7 Cell viability in Huh 7-Lunet (upper graph) and HeLa (lower graph) cell lines at increasing PEG2-b-PDAP concentration after 24 h (black bars), 48 h (stripped bars) and 72 h (grey bars) incubation times. *UTC: untreated controls. | ||
Besides being a well-known cytotoxic quinoline alkaloid discovered in 1966 by Wall and Wani,35 CPT was recently proposed as an effective antiviral against hepatitis C virus (HCV).29 Therefore, the capacity of PEG2-b-PDAP-CPT and PEG10-b-PDAP-CPT polymeric micelles to deliver CPT inside the cells and their ability to inhibit the replication of HCV virus in cells was essayed. The antiviral activity was assayed in Huh 5-2 cells (a variant of Lunet cells) at concentrations up to 0.1 mg mL−1. Under these conditions, the above mentioned cytotoxicity experiments indicated that the amphiphilic BCs do not strongly affect to the cell viability. Together with the antiviral activity, the cell viability of the culture was tested, so both effects of the CPT, cytotoxic and antiviral, could be quantified in cell culture. Because the BCs do not exhibit cytotoxic effect in the cells, the effect will be only due to the presence of CPT. Fig. 8 shows the results obtained for both CPT loaded polymeric micelles. HCV replicon replication rate and cell survival were evaluated at increasing CPT concentrations to determine 50% effective concentration (EC50) and 50% cytostatic concentration (CC50) and compared with values obtained for free CPT. The curves (line and symbol) represent the percentage of the initial virus replication rate that was inhibited during the experiment. The antiviral activity of PEG2-b-PDAP-CPT was very similar to free CPT, with EC = 0.015 μM, suggesting that this compound was released in the appropriate way in cell. In the case of PEG10-b-PDAP-CPT, the process of delivering the drug seemed to be slower, as EC50 was four fold higher in comparison with free CPT (0.06 and 0.015 μM, respectively) (Table 4).
 | ||
| Fig. 8 Inhibition of HCV replicon in Huh 5-2 cells. HCV replicon replication rate (curves) and cell survival (bars) for free CPT (stripped bars and triangles) and complexes (grey bars and circles). Upper graph, PEG2-b-PDAP-CPT; lower graph, PEG10-b-PDAP-CPT. *UTC: untreated controls. | ||
| Free CPT | PEG2-b-PDAP-CPT | PEG10-b-PDAP-CPT | |
|---|---|---|---|
| a Estimated values of EC50 (effective concentration 50%) and CC50 (cytotoxic concentration 50%) for the selected compounds in cell assays. | |||
| EC50 (μM) | 0.015 | 0.015 | 0.06 |
| CC50 (μM) | 0.5 | 0.4 | 0.5 |
| CC50 (μM)/EC50 (μM) | 34 | 27 | 8 |
In Fig. 8, cell viability is represented as bars. As CPT cytotoxic concentration (CC) was very high, no differences were found between free CPT and the CPT-loaded polymeric micelles. By calculating the CC/EC ratio (Table 4) it was observed that PEG2-b-PDAP-CPT value was quite similar to free CPT (around 30) but PEG10-b-PDAP-CPT value was lower (8) meaning a narrower therapeutic window.
Conclusions
The ability of polymer micelles obtained from amphiphilic block copolymer containing 2,6-diacylaminopyridine as nanocarriers for CPT delivery has been demonstrated. New amphiphilic polymers, constituted by hydrophilic PEG and hydrophobic 2,6-diacylaminopyridine based polymethacrylate of different molar mass but maintaining approximately the same hydrophilic/hydrophobic (35:75 wt/wt), have been prepared and can self-assemble forming spherical polymeric micelles having 18–25 nm diameter sizes. It has been demonstrated that the polymeric micelles, which are non-cytotoxic, can load CPT by simple diffusion up to 0.3–0.5 wt% drug contents while retaining their integrity and stability. According to the culture cells, the CPT-loaded polymeric micelles can efficiently deliver CPT inside the cells showing antiviral HVC activity. Therefore, this type of polymeric micelles might be used as carriers for drugs exhibiting difficulties to enter into cells.
When both described polymers are compared, polymeric micelles from the shortest polymer PEG2-b-PDAP, are sensitive to acidic media (they disintegrated due to protonation of the 2,6-diacylminopyridine unit). Also, the corresponding drug loaded micelles, PEG2-b-PDAP-CPT, are able to properly deliver CPT in cells. Therefore, it could be proposed as a potential drug carrier with a similar antiviral activity of free CPT.
Polymeric micelles from the largest polymer PEG10-b-PDAP are more stable at moderate acid media and also show lower antiviral activity, associated to a slower delivery of CPT. These characteristics might point to a more environmentally isolated internal core owing to the larger hydrophilic block length forming the outer shell when compared to PEG2-b-PDAP. This behavior together with the fact they encapsulate higher amounts of CPT than the shortest counterpart indicates that PEG10-b-PDAP might be interesting as a controlled release nanocarrier.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Acknowledgements
This work was supported by the MINECO, Spain (MAT2014-55205-P to LO and BFU2013-47064-P to AVC); Spanish Ministerio de Ciencia e Innovación (BFU2010-19451 to AVC); Spanish Ministerio de Educación, Cultura y Deporte (Grant BES-2013-064705 to AC and FPU13/3870 to RCG); Miguel Servet Program from Instituto de Salud Carlos III (CP07/00289 to OA and CPII13/0017), Fondo de Investigaciones Sanitarias (PI10/00186 and PI15/00663 to OA); FEDER funding; Diputación General de Aragón (Grant B136/13 to RCG, Digestive Pathology Group B01 to OA, Protein Targets Group B89 to AVC, E04 to MP/LO); Centro de Investigación Biomédica en Red en Enfermedades Hepáticas y Digestivas (CIBERehd) and Asociación Española de Gastroenterología (AEG). The authors would like to acknowledge the Servicios Generales de Apoyo a la Investigación – SAI and the Advanced Microscopy Laboratory – LMA of the Universidad de Zaragoza for the TEM observations. The authors additionally acknowledge the use of the CEQMA Services of the Universidad de Zaragoza-CSIC. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.References
- F. S. Bates and G. H. Fredrickson, Annu. Rev. Phys. Chem., 1990, 41, 525–557 CrossRef CAS PubMed.
- A. L. Larsen and E. M. Terentjev, Macromolecules, 2006, 39, 9497–9507 CrossRef CAS.
- A. L. Larsen and E. M. Terentjev, Macromolecules, 2006, 39, 9508–9518 CrossRef CAS.
- K. Letchford and H. Burt, Eur. J. Pharm. Biopharm., 2007, 65, 259–269 CrossRef CAS PubMed.
- T. Smart, H. Lomas, M. Massignani, M. V. Flores-Merino, L. R. Perez and G. Battaglia, Nano Today, 2008, 3, 38–46 CrossRef CAS.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- M. Elsabahy and K. L. Wooley, Chem. Soc. Rev., 2012, 41, 2545–2561 RSC.
- A. Rösler, G. W. M. Vandermeulen and H.-A. Klok, Adv. Drug Delivery Rev., 2012, 64, 270–279 CrossRef.
- J. Nicolas, S. Mura, D. Brambilla, N. Mackiewicz and P. Couvreur, Chem. Soc. Rev., 2013, 42, 1147–1235 RSC.
- M. Chen and M. Yin, Prog. Polym. Sci., 2014, 39, 365–395 CrossRef CAS.
- A. Kowalczuk, R. Trzcinska, B. Trzebicka, A. H. E. Müller, A. Dworak and C. B. Tsvetanov, Prog. Polym. Sci., 2014, 39, 43–86 CrossRef CAS.
- N. Kamaly, Z. Xiao, P. M. Valencia, A. F. Radovic-Moreno and O. C. Farokhzad, Chem. Soc. Rev., 2012, 41, 2971–3010 RSC.
- H. Wei, R.-X. Zhuo and X.-Z. Zhang, Prog. Polym. Sci., 2013, 38, 503–535 CrossRef CAS.
- J. Gong, M. Chen, Y. Zheng, S. Wang and Y. Wang, J. Controlled Release, 2012, 159, 312–323 CrossRef CAS PubMed.
- Y. Yan, G. K. Such, A. P. R. Johnston, J. P. Best and F. Caruso, ACS Nano, 2012, 6, 3663–3669 CrossRef CAS PubMed.
- Z. Ahmad, A. Shah, M. Siddiq and H.-B. Kraatz, RSC Adv., 2014, 4, 17028–17038 RSC.
- J. Ding, L. Chen, C. Xiao, L. Chen, X. Zhuang and X. Chen, Chem. Commun., 2014, 50, 11274–11290 RSC.
- H. Kuang, S. Wu, F. Meng, Z. Xie, X. Jing and Y. Huang, J. Mater. Chem., 2012, 22, 24832–24840 RSC.
- J. Fan, F. Zeng, S. Wu and X. Wang, Biomacromolecules, 2012, 13, 4126–4137 CrossRef CAS PubMed.
- H. Kuang, S. Wu, Z. Xie, F. Meng, X. Jing and Y. Huang, Biomacromolecules, 2012, 13, 3004–3012 CrossRef CAS PubMed.
- S. H. Kim, J. P. K. Tan, F. Nederberg, K. Fukushima, J. Colson, C. Yang, A. Nelson, Y.-Y. Yang and J. L. Hedrick, Biomaterials, 2010, 31, 8063–8071 CrossRef CAS PubMed.
- J. P. K. Tan, S. H. Kim, F. Nederberg, K. Fukushima, D. J. Coady, A. Nelson, Y. Y. Yang and J. L. Hedrick, Macromol. Rapid Commun., 2010, 31, 1187–1192 CrossRef CAS PubMed.
- S. Venkataraman, Z. A. Chowdhury, A. L. Lee, Y. W. Tong, I. Akiba and Y. Y. Yang, Macromol. Rapid Commun., 2013, 34, 652–658 CrossRef CAS PubMed.
- A. Concellón, E. Blasco, M. Piñol, L. Oriol, I. Díez, C. Berges, C. Sánchez-Somolinos and R. Alcalá, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 3173–3184 CrossRef.
- E. Blasco, J. del Barrio, C. Sánchez-Somolinos, M. Piñol and L. Oriol, Polym. Chem., 2013, 4, 2246–2254 RSC.
- V. Lohmann, F. Körner, J.-O. Koch, U. Herian, L. Theilmann and R. Bartenschlager, Science, 1999, 285, 110–113 CrossRef CAS PubMed.
- K. J. Blight, J. A. McKeating and C. M. Rice, J. Virol., 2002, 76, 13001–13014 CrossRef CAS PubMed.
- O. Abian, S. Vega, J. Sancho and A. Velazquez-Campoy, PLoS One, 2013, 8, e69773 CAS.
- R. Claveria-Gimeno, S. Vega, V. Grazu, J. M. de la Fuente, A. Lanas, A. Velazquez-Campoy and O. Abian, Int. J. Nanomed., 2015, 10, 2249–2259 CrossRef CAS PubMed.
- I. Jimenez-Pardo, R. Gonzalez-Pastor, A. Lancelot, R. Claveria-Gimeno, A. Velazquez-Campoy, O. Abian, M. B. Ros and T. Sierra, Macromol. Biosci., 2015, 15, 1381–1391 CrossRef CAS PubMed.
- E. A. Rainbolt, K. E. Washington, M. C. Biewer and M. C. Stefan, Polym. Chem., 2015, 6, 2369–2381 RSC.
- Y. Q. Yang, L. S. Zheng, X. D. Guo, Y. Qian and L. J. Zhang, Biomacromolecules, 2011, 12, 116–122 CrossRef CAS PubMed.
- U. Kedar, P. Phutane, S. Shidhaye and V. Kadam, Nanomedicine: Nanotechnology, Biology and Medicine, 2010, 6, 714–729 CrossRef CAS PubMed.
- M. E. Wall, M. C. Wani, C. E. Cook, K. H. Palmer, A. T. McPhail and G. A. Sim, J. Am. Chem. Soc., 1966, 88, 3888–3890 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Determination of average molar masses by 1H NMR and of the critical aggregation concentration (CAC) by fluorescence spectroscopy. See DOI: 10.1039/c6ra01714b |
| This journal is © The Royal Society of Chemistry 2016 |