
Structural and electronic properties of the PbCrO4 chrome yellow pigment and of its light sensitive sulfate-substituted compounds†
Abstract
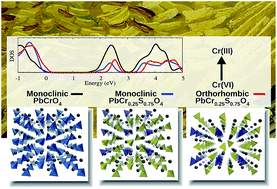
Chrome Yellows (CY) are a family of synthetic pigments of the formula PbCr(1−x)SxO4, used by van Gogh and other nineteenth-century masters, whose photo-degradation threatens the conservation of invaluable works of art. Experimental studies have demonstrated that the darkening of the pigment is provoked by the formation of superficial layers of Cr(III) oxides and sulfates due to the reduction of the native Cr(VI) to the Cr(III) oxidation state. A strong correlation between CY degradation and its chemical composition (sulfate richness) and structure (orthorhombic vs. monoclinic) was also experimentally put forward. Here we investigate a possible sulfate pathway to CY degradation, and investigate PbCr(1−x)SxO4 (x = 0, 0.25, 0.5, 0.75, 1) compounds in both orthorhombic and monoclinic phases by first principles DFT calculations. Our results show that the mixed PbCr(1−x)SxO4 and the native PbCrO4 share a similar electronic structure, though an energy up-shift of the conduction band is computed by both increasing the amount of sulfate and passing from the monoclinic to the orthorhombic phase. Our calculations suggests that, under a purely electronic picture, the Cr(VI) photo-reduction is more difficult for compounds with high sulfur concentration and an orthorhombic phase. Therefore, we conclude that degradation should be ascribed to other factors, such as different solubility and morphology.
 Please wait while we load your content...
Please wait while we load your content...

