DOI:
10.1039/C6RA03427F
(Paper)
RSC Adv., 2016,
6, 36327-36335
A comprehensive density functional theory study of the key role of fluorination and dual hydrogen bonding in the activation of the epoxide/CO2 coupling by fluorinated alcohols†
Received
5th February 2016
, Accepted 5th April 2016
First published on 6th April 2016
Abstract
The activation mechanism of the CO2/propylene oxide coupling catalysed by a bicomponent organocatalyst combining the use of TBABr with (multi)phenolic or fluorinated hydrogen bond donors (HBDs) was investigated using Density Functional Theory (DFT). Thus, it was shown that increasing the number of electron withdrawing trifluoromethyl substituents in HBDs strengthens their proton donor capability and allows a better stabilization by hydrogen bonding of the intermediates and transition states. In addition, the high efficiency of fluorinated monoalcohol activators is related to a dual hydrogen bonding mechanism by two fluorinated molecules that cooperatively contribute to the CO2/propylene oxide coupling.
Introduction
The synthesis of five-membered cyclic carbonates via the catalytic coupling of CO2 with epoxides is currently the subject of numerous studies.1–11 Although specific transition metal catalysts have proven to be efficient even under atmospheric pressure and ambient temperature,2,10,12 some of these complexes are highly sensitive to hydrolysis and oxidation, poorly selective and potentially toxic. To overcome these limitations, less/non-toxic and eco-friendly organocatalysts such as (functional) ionic liquids and halide salts have been extensively developed in the past years.13–26 If these organocatalysts allow the selective formation of cyclic carbonates from CO2 and epoxides, they usually display low activity and only show reasonable efficiency at high catalyst loading, at high pressure and high temperature (T > 100 °C) favoring their degradation.4,10,27–30 The catalytic activity of organocatalysts can be drastically enhanced by using suitable cocatalysts such as hydrogen bond donors (HBDs) which, as shown in several theoretical investigations, (i) interact with the O-donor group of epoxides facilitating the ring opening by the nucleophilic attack of the halide counter-anion of the halide salt or ionic liquid and, (ii) stabilize, by hydrogen bonding, the oxyanion formed after the epoxide ring-opening step.6,31–33 Phenolic derivatives,31,34,35 (amino)alcohols,36–38 fluorinated alcohols,39–41 carboxylic acids,42–44 lecithin,45 cellulose,46,47 chitosan,48–51 tannic acids52 and silanol63 have been proposed as potential hydrogen bond donors to fasten the CO2/epoxide coupling under mild experimental conditions. Among these HBDs, phenol derivatives, fluorinated alcohols and silanols were found to be the most efficient cocatalysts. Kleij et al. proposed a very efficient (multi)phenolic compound/ammonium iodide organocatalytic system that fastened the coupling of CO2 with epoxides at low temperature (25–45 °C) and pressure (1 MPa).31 Mattson et al. reported recently on the use of silanols as a new class of HBDs allowing the synthesis of cyclic carbonates at 0.1 MPa and 25 °C although the reaction rate was very low (18 h) despite the use of a very high catalyst (TBAI) and co-catalyst (silanol) loading (10 mol%).53 Recently, we reported a novel combination of onium salts with catalytic amount of fluorinated HBDs allowing the fast synthesis of cyclic carbonates within a few minutes under solvent free and mild experimental conditions.41
In a recent comparative study highlighting the cocatalytic activity of a series of commercially available HBDs, we demonstrated that fluorinated alcohols were the most efficient HBDs for the synthesis of cyclic carbonates from CO2 and epoxides under mild experimental conditions.39 From preliminary DFT calculations reported in ref. 41, we evidenced that two different HBDs diols (pyrogallol and 1,3-bis(2-hydroxyhexafluoroisopropyl)benzene) strongly reduced the relative energy of the reaction in comparison with the system without activator. A hydrogen-bond stabilization mechanism that impacted the energy of the epoxide ring-opening step was proposed. However, the origin of the high efficiency of fluorinated mono-alcohols in comparison with non-fluorinated (multi)phenolic activators was not understood. This prompted us to understand the origin of the unprecedented catalytic activity of fluorinated alcohols in comparison with other HBDs proposed in the literature. In this paper, we therefore report on the detailed mechanistic investigation, through molecular modeling, of the fluorinated HBDs/halide salts catalyzed CO2/propylene oxide coupling.
Experimental
Computational details
Preliminary calculations of equilibrium structures were performed using a semi-empirical model (AM1-D3H4)54,55 to determine the most stable conformations of the reactants, the catalysts and the products. These semi-empirical calculations were performed using the AMPAC software.56 Using the same semi-empirical model (AM1-D3H4), the CHAIN algorithm57 implemented in the AMPAC software was used for locating along the reaction path the conformations that were identified as intermediates and transition states. The lowest energy structures of the intermediates and transition states obtained at the AM1-D3H4 level were further investigated using the Density Functional Theory method (DFT) implemented in the Gaussian 09 package.58 DFT calculations of geometries, energies, and vibrational frequencies reported in this paper were carried out with the M06-2X functional59 using the 6-311G(d,p) basis set. All frequencies of each structure have been calculated to verify the presence of a single imaginary frequency for transition states and the absence of imaginary frequency for ground states. The intrinsic reaction coordinate (IRC) method has been used to verify that the obtained transition states were effectively connected to the desired minima. For all catalysts, a wide range of possible configurations and interactions have been modelled and the more stable of them are reported in this work. To consider entropic effects, the energies mentioned in this study correspond to the Gibbs free energy (ΔG).
Results and discussion
The mechanistic study through DFT simulation was realized for a model reaction involving the coupling of CO2 with propylene oxide (PO) catalyzed by tetrabutylammonium bromide (TBABr) in presence or not of HBDs. As previous reports60–64 figured out that the most probable pathway in the reaction mechanism involves the activation of the epoxide by the catalyst before addition of CO2, this activation mode was privileged in the present discussion. On the other hand, as it will be illustrated in this paper, the epoxide ring-opening step corresponds to the highest energy transition state and is assumed to be the Rate Determining Step (RDS), in line with previous reports.65,66 This is further supported by the following arguments:
(i) We consider that the initial energy of the reactants is conserved within the “system” along the reaction path even though the line dramatically dips down to some intermediates indicating a strong decrease in energy. Indeed, even if the energy leaves the reactants as they are transformed into an intermediate in an exergonic step, the released energy remains in solution and can be used by the intermediates to overcome their subsequent energy barriers.
(ii) When the reaction occurs in a solvent, a putative intermediate may only become a true detectable intermediate when the solvent fully stabilizes it. However, some intermediates might be “bypassed” because the dynamics of the solvent equilibration might be slower than the molecular reaction dynamics allowing the following elementary reaction step to occur before reaching the intermediate state of low energy.67 It is especially true for the systems studied in this work. Indeed, after the first elementary reaction, everything is in place to proceed until the formation of the end products. The activated intermediate (IR1; Fig. 1) is surrounded by CO2 molecules allowing a fast addition (compare to its stabilization) and the last step of the reaction (ring closure) is an intramolecular rearrangement which is fast as the “excess” energy is already available in the degrees of freedom involved in the reaction coordinate of this final step. The “bypass” of low energy intermediates is supported by the fact that we do not experimentally detect (in our in situ kinetic studies) the very low energy intermediates reported in our DFT study.
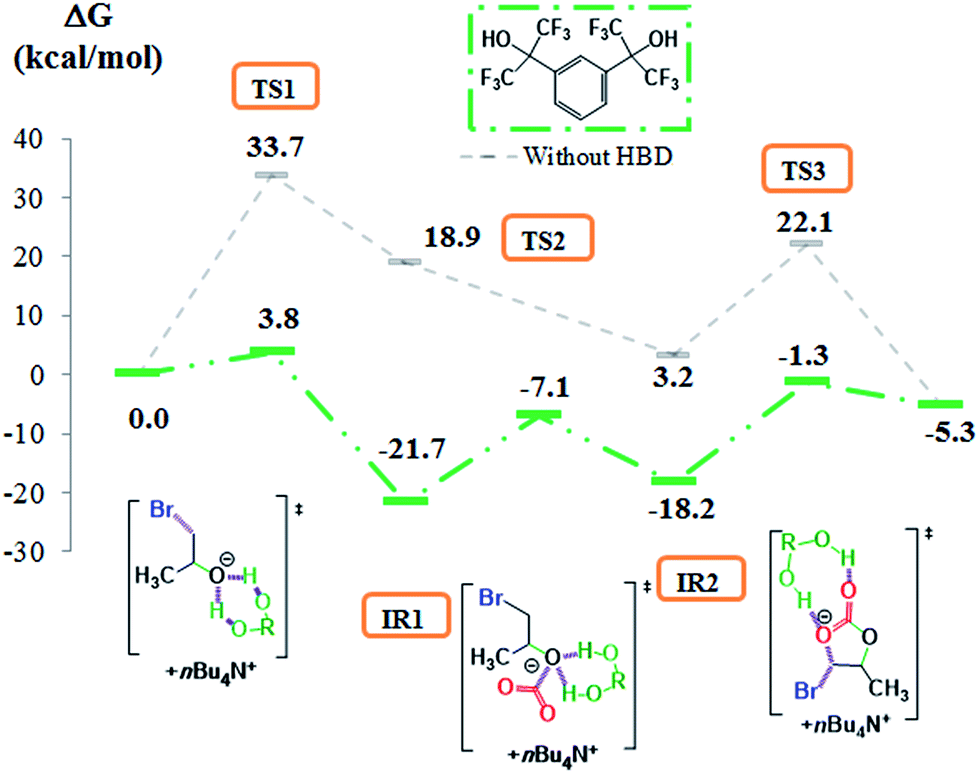
 |
| | Fig. 1 Free energy profiles (M06-2X/6-311G(d,p)) and Lewis structures for the propylene oxide/CO2 coupling catalyzed by TBABr. | |
Considering this discussion, the first step of the reaction, thus the ring-opening of the epoxide, is the RDS, whatever the catalytic system investigated.
CO2/PO coupling without HBDs
Schematic potential energy profiles obtained at the M06-2X/6-311G(d,p) level and schematic Lewis structures for the CO2/PO coupling catalyzed by TBABr are depicted in Fig. 1. After having fully optimized the TBABr, PO, propylene carbonate (PC) and CO2 structures, the overall cycloaddition pathway between PO and CO2 has been modelled by a three-step mechanism. The initial state corresponds to the sum of the free energies of each compound (PO, CO2, TBABr) which is taken as the zero energy. The first transition state TS1 corresponds to the nucleophilic attack of the epoxide by the bromine ion of the organocatalyst inducing the opening of the C–O–C bond (ESI1†) and the stabilization of the resulting oxy-anion by the alkyl chains of TBA+ by van der Waals interactions. The nucleophilic attack on the secondary carbon of PO was only considered as it was reported to be the most favourable.64 Then, a bromohydrin ion corresponding to the intermediate IR1 is obtained by formation of a C–Br bond between Br− and the non-substituted carbon atom of the epoxide. As IR1 is not stabilized by strong interactions, the insertion of CO2 onto the oxy-anion occurs without the existence of a transition state (TS2) and leads to a more stable bromo-alkylcarbonate intermediate (IR2) that is stabilized by van der Waals interactions between the carbonate ion and the alkyl chains of TBA+. Finally, a second transition step (TS3), which consists in the breaking of the C–Br bond and the intramolecular ring-closure of the intermediate IR2, provides propylene carbonate (PC) while regenerating the ammonium salt. Note that this “backbiting” step is named TS3 instead of TS2 for an easier comparison of the reaction pathway with that obtained using hydrogen bond activators (see below). From the DFT calculations, the formation of the first transition state (TS1) corresponds to the rate-determining step and 33.7 kcal mol−1 are required to obtain propylene carbonate with TBABr. In view of this high activation energy value, the energy barrier is too high for the non-activated CO2/PO coupling to happen under mild conditions. Thus, efficient activators able to significantly decrease the energy barrier for the formation of TS1 have to be identified.
CO2/PO coupling using (multi)phenolic HBDs
(Multi)phenolic derivatives were reported amongst the most efficient HBDs for the TBAI catalyzed coupling of CO2 with epoxides.43,47 In Kleij's study, the beneficial role of such HBDs that activate the epoxide and reduce the energy barrier of the limiting step was highlighted through a detailed DFT study. However, the presence of the TBA+ counter-ion was not taken into account in the initial study. For coherence with our DFT results, we reinvestigated the overall reaction pathway for which the energy profiles are depicted in Fig. 2 for phenol (blue), pyrocatechol (magenta) and pyrogallol (red). The sum of free energies of isolated reactants (CO2, PO) and catalysts (TBABr, phenol derivative) is taken as zero. As reported by Kleij, whatever the phenolic HBD, a three-step mechanism (instead of two when TBABr is used as sole catalyst) whose energy depends on the nature of the phenol derivative is observed. As the mechanisms are similar for all (multi)phenolic derivatives, the reaction pathway will be only described for pyrogallol (Fig. 3). In a first step, the nucleophilic Br− anion attacks the less-substituted C atom of PO leading to the epoxide ring-opening (TS1). Pyrogallol and TBA+ have a huge impact on the stabilization of TS1 via the formation of hydrogen bonds and van der Waals interactions. Indeed, the central OH group of the pyrogallol transfers its proton to the bromohydrin (1.47 Å) creating an O–H bond. The first vicinal hydroxyl function allows for the stabilization of the O atom of the bromohydrin ion (1.78 Å) and the second one stabilizes the central O atom of pyrogallol (2.04 Å), consistently with Kleij's observations. However, the presence of the TBA+ ion induces a new stabilization by covering the complex via creation of van der Waals interactions with the aromatic ring of pyrogallol. This overall stabilization mechanism decreases importantly the energy of the first step compared to the pathway described previously without HBD. From TS1, the formation of the bromohydrin and the phenoxide anion leads to the stable intermediate IR1. In a second step, the C atom of the CO2 approaches the O atom of the bromohydrin implying the breaking of the O–H bond previously created (TS2). The bromo-alkylcarbonate (IR2) is formed by the formation of a bond between CO2 and the bromohydrin. IR2 is stabilized through a similar mechanism that the one discussed for the formation of IR1. The final step involves a torsional deformation of the bromo-alkylcarbonate and the C–Br bond rupture (TS3). PC is formed by ring-closure of the alkylcarbonate with the release of TBABr and pyrogallol. The relative energy of this last path is drastically reduced in comparison with the one reported without HBD thanks to the hydrogen-bonding stabilization mechanism driven by pyrogallol. From the DFT studies, the number of vicinal hydroxyl functions of the phenolic HBD has an important influence on the energy of this path (Fig. 2) as the required Gibbs energies to overcome the energy barrier are 7.7 kcal mol−1, 10.5 kcal mol−1 and 16.6 kcal mol−1 for pyrogallol, pyrocatechol and phenol, respectively, which is much lower than the value determined when TBABr is the sole catalyst (ΔGTS1 = 33.7 kcal mol−1). If the general trends, i.e. the mechanism and the decrease of the activation energies, are fully consistent with Kleij's experiment, the Gibbs energies values determined in our study cannot be compared with the values mentioned in Kleij's work as the functional and the basis set that we selected are different.
 |
| | Fig. 2 Free energy profiles (M06-2X/6-311G(d,p)) and Lewis structures of the transition states for CO2/PO coupling catalyzed by TBABr (grey), TBABr plus phenol (blue), TBABr plus pyrocatechol (magenta) and TBABr plus pyrogallol (red). | |
 |
| | Fig. 3 Optimized geometries of the structures (M06-2X/6-311G(d,p)) of the CO2/PO coupling, in the presence of pyrogallol/TBABr as catalysts. The dashed lines depict hydrogen bond interactions and intermolecular distances are given in angstrom. | |
CO2/PO coupling using fluorinated monoalcohols as HBDs
Recently, we reported that some fluorinated alcohols show very high cocatalytic activity for the TBABr promoted coupling of CO2 with epoxides and even proved to be more efficient than (multi)phenolic HBDs under similar experimental conditions.39,41 The reaction mechanism was then studied in detail and the energy profiles were reported on Fig. 4 for fluorinated monoalcohols, i.e. trifluoroethanol (TFE, yellow), hexafluoro-tert-butanol (HFB, purple), perfluoro-tert-butanol (PFB, light blue) and hexafluoro-(p-tolyl)-isopropanol (HFTI, orange). For illustration, the reaction pathway describing the structure of all intermediates and transitions states is reported in detail for perfluoro-tert-butanol in ESI2† where the hydrogen-bonding stabilization mechanism driven by the activator and the van der Waals interactions with TBA+ are clearly identified at each step of the reaction. Similarly to phenolic HBDs, we observe in Fig. 4 that for all fluorinated alcohols, the reaction answers the three-step mechanism whose energy depends on the nature of the HBD. Whatever the fluorinated activator, the relative energy of the elementary paths, and more particularly regarding the energy of the epoxide ring-opening step, is significantly reduced in comparison with that reported without activator. Fig. 4 also highlights the positive influence of the increasing number of trifluoromethyl substituents in HBDs. Indeed, the Gibbs energy decreases in the order TFE (1 CF3) > HFB (2 CF3) > PFB (3 CF3). As fluorine atom is an electronegative element, the Natural Bond Orbital (NBO) analysis of TS1 reveals that the positive charge (qH) on the proton of the hydroxyl function increases with the number of electron withdrawing trifluoromethyl groups (Table 1). To further ascertain the positive role of the trifluoromethyl groups, the substitution of one CF3 of PFB by a less attractive p-tolyl group (HFTI) induces a slight increase of the Gibbs energy required for the CO2/PO coupling, which value is intermediate to the ones reported for PFB and HFB. On the other hand, for the first transition state (TS1), the O⋯H distance between the O atom of the opened epoxide and the proton of the fluorinated alcohol decreases when the charge determined by the NBO analysis on the H atom increases (Table 1) highlighting stronger hydrogen bond interactions. Thus, the higher the hydrogen bond interaction between the opened epoxide and the fluorinated alcohol is, the lower the Gibbs energy is required for the first reaction step as confirmed by the Gibbs energy values determined for TFE (ΔG = 20.3 kcal mol−1), HFB (ΔG = 15.8 kcal mol−1), HFTI (ΔG = 14.4 kcal mol−1) and PFB (ΔG = 12.0 kcal mol−1). Although the Gibbs energy corresponding to the first limiting step of the CO2/PO coupling using PFB (ΔG = 12.0 kcal mol−1) is significantly lower than with phenol (ΔG = 16.6 kcal mol−1), it remains higher than the value determined for pyrogallol (ΔG = 7.7 kcal mol−1). This observation seems to contradict our experimental results that showed that fluorinated HBDs were better activators than the (multi)phenolic ones.39
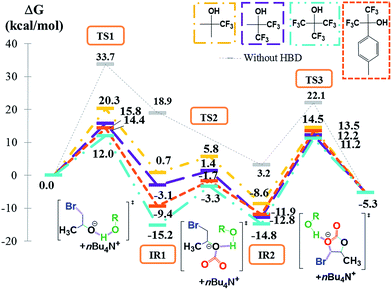 |
| | Fig. 4 Free energy profile (M06-2X/6-311G(d,p)) and Lewis structures of transition states for the CO2/PO coupling catalyzed by TBABr (grey), TBABr plus TFE (yellow), TBABr plus HFB (magenta) TBABr plus PFB (blue) and TBABr plus HFTI (orange). | |
Table 1 Charge on the proton of the (fluorinated) alcohol obtained by Natural Bond Orbital (NBO) analysis and O⋯H length between the epoxide and the (fluorinated) alcohol depending on the structure of the (fluorinated) alcohol for the transition state resulting from the nucleophilic attack of the bromine anion on the epoxide ring (TS1)
| Fluorinated alcohol |
qH (me) |
–O⋯H–O– length (Å) |
| Tert-butanol |
0.45 |
1.70 |
| Trifluoroethanol |
0.46 |
1.44 |
| Hexafluoro-tert-butanol |
0.47 |
1.33 |
| Hexafluoro-(p-tolyl)-isopropanol |
0.48 |
1.26 |
| Perfluoro-tert-butanol |
0.49 |
1.09 |
To understand the origin of this disagreement, we supposed that dual hydrogen bonding activation mechanism involving two fluorinated monoalcohols could be responsible for an enhanced stabilization of the transition states and the intermediates. Indeed, Berkessel et al. reported that the boosting catalytic effect of fluorinated alcohols, used as solvents for the olefins epoxidation, was due to their aggregation and the formation of a multiple H-bond network.68,69 This assumption is also further supported by our previous NMR titrations experiments that highlighted the formation of complexes between the epoxide and more than one molecule of fluorinated HBD.41 Both the reaction mechanism and the Gibbs energy were then investigated for the dual hydrogen bonding activation with TFE, HFB, HFTI and PFB (Fig. 5). As shown in Fig. 6 (for PFB), two molecules participate to the stabilization of the O atom of the epoxide via H-bonds. Due to the steric hindrance caused by the methyl substituent of PO, the structure of TS1 exhibits two asymmetric H-bonds (1.48 Å and 1.57 Å). For sake of comparison, DFT calculations on ethylene oxide provide a complete symmetric system with two identical H-bonds lengths. The resulting IR1 is characterized by a PFB–bromohydrin–PFB H-bonds sequence for which the O atom of the bromohydrin occupies a central position of the “IR1 complex”. In TS2, both PFB molecules play a different role. One interacts with one O atom of the former CO2 while the second interacts with the oxygen of the former epoxide. Then, each PFB molecules stabilizes one terminal O atom of the carbonate group by hydrogen bonding until the formation of the cyclic carbonate (IR2 and TS3 on Fig. 6). Besides these mechanistic considerations, the dual hydrogen bonding activation by two fluorinated monoalcohols impacts significantly the Gibbs energy profile (Fig. 5) compared to the one with only one fluorinated alcohol molecule (Fig. 4). This is clearly evidenced for the first – and limiting – step as the Gibbs energy is decreased by 6.6, 4.9, 4.7 and 8.3 kcal mol−1 for TFE, HFB, HFTI and PFB, respectively. Thus, these DFT calculations highlight the origin of the high cocatalytic activity of fluorinated monoalcohol thanks to a dual hydrogen bonding activation by two fluorinated monoalcohols that cooperatively contribute to the coupling of CO2 with PO.
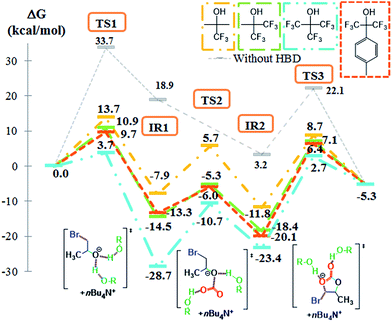 |
| | Fig. 5 Free energy profiles (M06-2X/6-311G(d,p)) and Lewis structures of the transition states for the CO2/PO coupling catalyzed by TBABr (grey), TBABr plus two molecules of TFE (yellow), TBABr plus two molecules of HFB (green) TBABr plus two molecules of HFTI (orange) and TBABr plus two molecules of PFB (blue). | |
 |
| | Fig. 6 Optimized geometries (M06-2X/6-311G(d,p)) of the structures of the CO2/PO coupling in the presence of TBABr plus two molecules of PFB as catalysts. The dashed lines depict the intermolecular distances in angstrom. | |
CO2/PO coupling using fluorinated HBDs: the key role of the CF3 groups
To determine to which extent the substitution of a methyl group by a trifluoromethyl one impacts the capability of HBD to generate strong hydrogen bonding and efficient stabilization of the transition states and intermediates, we compared the Gibbs energy profiles of the TBABr promoted coupling of CO2 with PO using two molecules of tert-butanol or PFB as cocatalysts. From Fig. 7, we observe that the relative energy of each transition states and intermediates is drastically reduced in the presence of PFB in comparison with the values reported for tert-butanol. Clearly, the non-fluorinated activator has a poor effect on the stabilization of both the intermediates and transitions states that results from its lower capability to generate strong hydrogen bonds. Indeed, if the modelled structures of transition states TS1 (epoxide ring-opening) and TS3 (carbonate ring closure) with tert-butanol and PFB are similar (Fig. 6 and ESI3†), the hydrogen bond length between the proton of the HBDs and the reactive center are different. For TS1, the distances between the protons of the hydroxyl group and the O atom of the ring-opened epoxide are much longer for tert-butanol (1.70 Å and 1.74 Å, ESI3†) than for PFB (1.48 Å and 1.57 Å, Fig. 6). The NBO analysis of both the TS1 reveals that the charge distribution is dissimilar. The atomic charges of the proton of the hydroxyl functionalities are 0.517 and 0.512 in the case of tert-butanol against 0.533 and 0.522 for PFB, in line with the more acidic character of PFB. Therefore, the stabilization of the resulting bromohydrin IR1 is significantly weaker with tert-butanol and the proton transfer to the bromohydrin does not occur. This explains the significant difference in the Gibbs energy values of TS1 using tert-butanol (ΔG = 19.9 kcal mol−1) or PFB (ΔG = 3.7 kcal mol−1) as HBDs. The same marked difference exists between 1,3-bis(2-hydroxyhexafluoroisopropyl)-benzene (1,3-bis-HFAB) and its non-fluorinated counterpart (α,α׳-dihydroxy-1,3-diisopropylbenzene, ESI4†).
 |
| | Fig. 7 Free energy profiles (M06-2X/6-311G(d,p)) and Lewis structures of the transition states for the CO2/PO coupling catalyzed by TBABr plus two molecules of tert-butanol (dark blue) and TBABr plus two molecules of PFB (light blue). | |
CO2/PO coupling using fluorinated diols as HBDs
In our previous work, we found that a fluorinated double hydrogen bond donor, i.e. 1,3-bis-HFAB, an alcohol bearing two hexafluoroisopropyl groups, showed an impressive cocatalytic activity for the TBABr promoted coupling of CO2 with epoxides. The origin of this cocatalytic activity was elucidated by DFT calculation. Fig. 8 reports on the detailed reaction mechanism answering the three elementary step pathway. The molecular structures of the corresponding intermediates and transition states are displayed in Fig. 9. To maximize clarity of the figure, the TBA+ cation which stabilizes the system by van der Waals interactions was represented using a skeleton model. First the nucleophilic attack of the bromine ion onto the non-substituted carbon atom of PO leads to the epoxide ring-opening. The resulting negatively charged O atom of the transition state (TS1) is stabilized by both hydroxyl functional groups of 1,3-bis-HFAB (1.44 Å and 1.92 Å). In addition, a weak C–H⋯O hydrogen bond interaction is formed between the O atom of the bromohydrin anion and the aromatic proton in ortho position of hexafluoro alcohol functionalities (2.32 Å) that contributes cooperatively with the two O–H⋯O hydrogen bonds to the stabilization of TS1. The intermediate IR1 is obtained after the formation of the C–Br bond and the proton transfer from 1,3-bis-HFAB to the O atom. Consequently, the bromohydrin is stabilized by the second hydroxyl function of the hydrogen-bond donor (1.82 Å). Then, the attack of CO2 by the O atom of the bromohydrin implies a concomitant breaking of the O–H bond previously created (TS2). Hydrogen-bonds are formed between 1,3-bis-HFAB and the bromo-alkylcarbonate leading to a stable intermediate (IR2). Finally, the torsional deformation of the bromo-alkylcarbonate occurs and both hydroxyl functions and the aromatic proton of 1,3-bis-HFAB stabilize the two O atoms of the inserted carbon dioxide (TS3). The concerted C–Br bond rupture and the carbonate ring-closing lead to the formation of propylene carbonate. The Gibbs energies required for the epoxide ring-opening and the carbonate ring-closing steps are significantly lower than that calculated with pyrogallol as activator. In particular, only 3.8 kcal mol−1 are necessary for the CO2/PO coupling (Fig. 8) instead of 7.7 kcal mol−1 in the case of pyrogallol (Fig. 2). These calculations are consistent with the experimental data showing that 1,3-bis-HFAB is a more efficient activator than pyrogallol.39 This higher efficiency of 1,3-bis-HFAB might be related to the stabilization mechanism that involves three intermolecular H-bonds (see Fig. 9-TS1) instead of only two for pyrogallol (see Fig. 3-TS1). These results clearly highlight that the number of hydrogen atoms able to participate to the activation of epoxides and their ability to stabilize the transitions states and intermediates by hydrogen bonding are key parameters for optimizing the activity of the cocatalysts.
 |
| | Fig. 8 Free energy profiles (M06-2X/6-311G(d,p)) and Lewis structures of the transition states for the CO2/PO coupling catalyzed by TBABr (grey) and TBABr plus 1,3-bis-HFAB (green). | |
 |
| | Fig. 9 Optimized geometries of the structures (M06-2X/6-311G(d,p)) of the CO2/PO coupling in the presence of 1,3-bis-HFAB/TBABr as catalysts. The dashed lines depict hydrogen bond interactions and intermolecular distances are given in angstrom. | |
Conclusions
A detailed mechanistic investigation by DFT calculations of the TBABr promoted coupling of CO2 with propylene oxide using a series of hydrogen bond donors (HBDs) activators is reported. In particular, we have investigated phenol derivatives and fluorinated alcohols that have shown unprecedented catalytic activity in comparison with other HBDs proposed in the literature. Thus, we demonstrated how the substitution of alcohols with trifluoromethyl groups significantly improves the stabilization of transition states and the intermediates generated during the reaction by a significant decrease of the Gibbs energy of each step and especially, the one corresponding to the ring-opening of the epoxide. The results reveal that the trifluoromethyl groups strongly influences the charge distribution of HBDs and, in particular, the acidity of the hydroxyl groups that strengthens hydrogen-bond interactions. Indeed, the Gibbs energy decreases in the order TFE (1 CF3) > HFB (2 CF3) > PFB (3 CF3). We also showed that for single HBDs, the stabilization of the transition states and intermediates was further improved in the presence of two molecules of fluorinated monoalcohols that contribute cooperatively to the catalytic mechanism through a dual hydrogen bonding activation. Finally, DFT calculations highlight that the double HBD, 1,3-bis-HFAB, is a better cocatalyst than pyrogallol, one of the most efficient (multi)phenolic activators, because three intermolecular H-bonds are involved with 1,3-bis-HFAB instead of only two in the case of pyrogallol.
Acknowledgements
The PhD fellowship of M. ALVES was co-funded by the “IDEX (Initiative d’Excellence) program of the University of Bordeaux” and the “University of LIEGE” through the international doctoral school program “IDS-FunMat” supported by the European Community (ERASMUS MUNDUS Doctoral program). We also thank computational facilities provided by the ‘‘Pôle Modélisation’’ of the ISM and the MCIA (Mesocentre de Calcul Intensif Aquitain) of the University of Bordeaux (http://www.mcia.univ-bordeaux.fr) financed by the ‘‘Conseil Regional d’Aquitaine’’ and the French Ministry of Research and Technology. The authors of Liège thank the “Region Wallonne” in the frame of the CO2Green, Sinopliss and Flycoat projects, the "Belgian Science Policy" in the frame of the "Interuniversity Attraction Poles Programme (IAP VII/5) – Functional Supramolecular Systems" and the "Fonds National pour la Recherche Scientifique” (F.R.S.-FNRS) for financial supports. C.D. is Research Director by F.R.S.-FNRS.
Notes and references
- M. Aresta, Carbon Dioxide as Chemical Feedstock, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2010 Search PubMed.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC.
- T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- P. P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2, 2169–2187 CAS.
- A. Decortes, A. M. Castilla and A. W. Kleij, Angew. Chem., Int. Ed., 2010, 49, 9822–9837 CrossRef CAS PubMed.
- J.-Q. Wang, K. Dong, W.-G. Cheng, J. Sun and S.-J. Zhang, Catal. Sci. Technol., 2012, 2, 1480–1484 CAS.
- D. J. Darensbourg and S. J. Wilson, Green Chem., 2012, 14, 2665–2671 RSC.
- C. J. Whiteoak and A. W. Kleij, Synlett, 2013, 24, 1748–1756 CrossRef CAS.
- Z.-Z. Yang, L.-N. He, J. Gao, A.-H. Liu and B. Yu, Energy Environ. Sci., 2012, 5, 6602–6639 CAS.
- C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef.
- A. H. Liu, Y. N. Li and L. N. He, Pure Appl. Chem., 2012, 84, 581–602 CrossRef CAS.
- M. North and C. Young, Catal. Sci. Technol., 2011, 1, 93–99 CAS.
- J. Sun, S. I. Fujita and M. Arai, J. Organomet. Chem., 2005, 690, 3490–3497 CrossRef CAS.
- T. Seki, J. D. Grunwaldt and A. Baiker, J. Phys. Chem. B, 2009, 113, 114–122 CrossRef CAS PubMed.
- J. Li, L. Wang, F. Shi, S. Liu, Y. He, L. Lu, X. Ma and Y. Deng, Catal. Lett., 2011, 141, 339–346 CrossRef CAS.
- Z. Z. Yang, L. N. He, C. X. Miao and S. Chanfreau, Adv. Synth. Catal., 2010, 352, 2233–2240 CrossRef CAS.
- H. Y. Ju, M. D. Manju, K. H. Kim, S. W. Park and D. W. Park, J. Ind. Eng. Chem., 2008, 14, 157–160 CrossRef CAS.
- S. Foltran, J. Alsarraf, F. Robert, Y. Landais, E. Cloutet, H. Cramail and T. Tassaing, Catal. Sci. Technol., 2013, 3, 1046–1055 CAS.
- W.-L. Wong, L. Y. S. Lee, K.-P. Ho, Z.-Y. Zhou, T. Fan, Z. Lin and K.-Y. Wong, Appl. Catal., A, 2014, 472, 160–166 CrossRef CAS.
- D. Wei-Li, J. Bi, L. Sheng-Lian, L. Xu-Biao, T. Xin-Man and A. Chak-Tong, Catal. Today, 2014, 233, 92–99 CrossRef.
- J. Sun, S. Zhang, W. Cheng and J. Ren, Tetrahedron Lett., 2008, 49, 3588–3591 CrossRef CAS.
- W. L. Dai, J. Bi, S. L. Luo, X. B. Luo, X. M. Tu and C. T. Au, Catal. Sci. Technol., 2014, 4, 556–562 CAS.
- O. Coulembier, S. Moins, V. Lemaur, R. Lazzaroni and P. Dubois, J. CO2 Util., 2015, 10, 7–11 CrossRef CAS.
- J. Tharun, G. Mathai, R. Roshan, A. C. Kathalikkattil, K. Bomi and D.-W. Park, Phys. Chem. Chem. Phys., 2013, 15, 9029–9033 RSC.
- A. J. R. Amaral, J. F. J. Coelho and A. C. Serra, Tetrahedron Lett., 2013, 54, 5518–5522 CrossRef CAS.
- M. H. Anthofer, M. E. Wilhelm, M. Cokoja, M. Drees, W. A. Herrmann and F. E. Kühn, ChemCatChem, 2015, 7, 94–98 CrossRef CAS.
- J. Łukaszczyk, K. Jaszcz, W. Kuran and T. Listos, Macromol. Rapid Commun., 2000, 21, 754–757 CrossRef.
- D. J. Darensbourg and W.-C. Chung, Macromolecules, 2014, 47, 4943–4948 CrossRef CAS.
- S. Kumar, S. L. Jain and B. Sain, Catal. Lett., 2012, 142, 615–618 CrossRef CAS.
- G. Fiorani, W. Guo and A. W. Kleij, Green Chem., 2015, 17, 1375–1389 RSC.
- C. J. Whiteoak, A. Nova, F. Maseras and A. W. Kleij, ChemSusChem, 2012, 5, 2032–2038 CrossRef CAS PubMed.
- J. Ma, J. Liu, Z. Zhang and B. Han, Green Chem., 2012, 14, 2410–2420 RSC.
- L. Wang, X. Jin, P. Li, J. Zhang, H. He and S. Zhang, Ind. Eng. Chem. Res., 2014, 53, 8426–8435 CrossRef CAS.
- J. W. Huang and M. Shi, J. Org. Chem., 2003, 68, 6705–6709 CrossRef CAS PubMed.
- Y. M. Shen, W. L. Duan and M. Shi, Eur. J. Org. Chem., 2004, 3080–3089 CrossRef CAS.
- T. Werner, N. Tenhumberg and H. Büttner, ChemCatChem, 2014, 6, 3493–3500 CrossRef CAS.
- M. E. Wilhelm, M. H. Anthofer, M. Cokoja, I. I. E. Markovits, W. A. Herrmann and F. E. Kühn, ChemSusChem, 2014, 7, 1357–1360 CrossRef CAS PubMed.
- T. Werner and N. Tenhumberg, J. CO2 Util., 2014, 7, 39–45 CrossRef CAS.
- M. Alves, B. Grignard, S. Gennen, R. Mereau, C. Detrembleur, C. Jerome and T. Tassaing, Catal. Sci. Technol., 2015, 5, 4636–4643 CAS.
- M. Alves, B. Grignard, S. Gennen, C. Detrembleur, C. Jerome and T. Tassaing, RSC Adv., 2015, 5, 53629–53636 RSC.
- S. Gennen, M. Alves, R. Méreau, T. Tassaing, B. Gilbert, C. Detrembleur, C. Jerome and B. Grignard, ChemSusChem, 2015, 8, 1845–1849 CrossRef CAS PubMed.
- W. Cheng, Z. Fu, J. Wang, J. Sun and S. Zhang, Synth. Commun., 2012, 42, 2564–2573 CrossRef CAS.
- L. Han, H.-J. Choi, S.-J. Choi, B. Liu and D.-W. Park, Green Chem., 2011, 13, 1023–1028 RSC.
- J. Tharun, G. Mathai, A. C. Kathalikkattil, R. Roshan, J.-Y. Kwak and D.-W. Park, Green Chem., 2013, 15, 1673–1677 RSC.
- J. Song, B. Zhang, P. Zhang, J. Ma, J. Liu, H. Fan, T. Jiang and B. Han, Catal. Today, 2012, 183, 130–135 CrossRef CAS.
- S. Liang, H. Liu, T. Jiang, J. Song, G. Yang and B. Han, Chem. Commun., 2011, 47, 2131–2133 RSC.
- J. Sun, W. Cheng, Z. Yang, J. Wang, T. Xu, J. Xin and S. Zhang, Green Chem., 2014, 16, 3071–3078 RSC.
- J. Sun, J. Wang, W. Cheng, J. Zhang, X. Li, S. Zhang and Y. She, Green Chem., 2012, 14, 654–660 RSC.
- J. Tharun, Y. Hwang, R. Roshan, S. Ahn, A. C. Kathalikkattil and D. W. Park, Catal. Sci. Technol., 2012, 2, 1674–1680 CAS.
- Y. Zhao, J. S. Tian, X. H. Qi, Z. N. Han, Y. Y. Zhuang and L. N. He, J. Mol. Catal. A: Chem., 2007, 271, 284–289 CrossRef CAS.
- C. Jing-Xian, J. Bi, D. Wei-Li, D. Sen-Lin, C. Liu-Ren, C. Zong-Jie, L. Sheng-Lian, L. Xu-Biao, T. Xin-Man and A. Chak-Tong, Appl. Catal., A, 2014, 484, 26–32 CrossRef.
- S. Sopeña, G. Fiorani, C. Martín and A. W. Kleij, ChemSusChem, 2015, 8, 3179 CrossRef PubMed.
- A. M. Hardman-Baldwin and A. E. Mattson, ChemSusChem, 2014, 7, 3275–3278 CrossRef CAS PubMed.
- M. J. S. Dewar, E. G. Zoebisch, E. F. Healy and J. J. P. Stewart, J. Am. Chem. Soc., 1985, 107, 3902–3909 CrossRef CAS.
- J. Řezáč and P. Hobza, J. Chem. Theory Comput., 2012, 8, 141–151 CrossRef PubMed.
- AMPAC 10, ©, Semichem, Inc, PO Box 1649, Shawnee, KS 66222, 1992–2013 Search PubMed.
- D. A. Liotard, Int. J. Quantum Chem., 1992, 44, 723–741 CrossRef CAS.
- M. J. Frisch and A. D. Becke, Gaussian 09, Revision A.02, 2009, 98, 5648–5652– Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- J.-Q. Wang, J. Sun, W.-G. Cheng, K. Dong, X.-P. Zhang and S.-J. Zhang, Phys. Chem. Chem. Phys., 2012, 14, 11021–11026 RSC.
- H. Sun and D. Zhang, J. Phys. Chem. A, 2007, 111, 8036–8043 CrossRef CAS PubMed.
- S. Marmitt and P. F. B. Gonçalves, J. Comput. Chem., 2015, 36, 1322–1333 CrossRef CAS PubMed.
- Y. Ren, C. H. Guo, J. F. Jia and H. S. Wu, J. Phys. Chem. A, 2011, 115, 2258–2267 CrossRef CAS PubMed.
- S. Foltran, R. Mereau and T. Tassaing, Catal. Sci. Technol., 2014, 4, 1585–1597 CAS.
- S. Kozuch and J. M. Martin, ChemPhysChem, 2011, 12, 1413–1418 CrossRef CAS PubMed.
- E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science, Sausalito, 2004 Search PubMed.
- Z. Chen, Y. Nieves-Quinones, J. R. Waas and D. A. Singleton, J. Am. Chem. Soc., 2014, 136, 13122–13125 CrossRef CAS PubMed.
- A. Berkessel, J. A. Adrio, D. Hüttenhain and J. M. Neudörfl, J. Am. Chem. Soc., 2006, 128, 8421–8426 CrossRef CAS PubMed.
- A. Berkessel and J. A. Adrio, J. Am. Chem. Soc., 2006, 128, 13412–13420 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra03427f |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.