
Structural analogues of Hoveyda–Grubbs catalysts bearing the 1-benzofuran moiety or isopropoxy-1-benzofuran derivatives as olefin metathesis catalysts†
B. Trzaskowski*a and
K. Ostrowskab
aCentre of New Technologies, University of Warsaw, 02-097 Warszawa, Poland. E-mail: trzask@cent.uw.edu.pl
bFaculty of Pharmacy, Medical University of Warsaw, 02-097 Warszawa, Poland
First published on 18th February 2016
Abstract
We have used the DFT/M06-D3 computational method to study structures and activation free energies for a series of Hoveyda–Grubbs-like catalysts with the isopropoxybenzene part replaced by 1-benzofuran and ten derivatives of isopropoxy-1-benzofuran. We show that while Ru coordination by the benzofuran oxygen atom is relatively strong, simple modification of isopropoxybenzene by benzofuran doesn't lead to a stable ruthenium complex due to geometrical reasons. Some of the more complex modifications, which replace the phenyl group of the isopropoxybenzylidene with the benzofuran core but leave the isopropoxy group intact, are good candidates for potent metathesis catalysts with free energies of activation of the dissociative path 2–5 kcal mol−1 lower than the Hoveyda–Grubbs catalyst.
Introduction
Since their discovery in 1999, second-generation Grubbs complexes and their modifications suggested by Hoveyda (1) have rapidly evolved into a large family of catalysts with varying properties.1 These catalysts have been the key to widespread applications of olefin metathesis in organic synthesis, and allowed important applications that extend to a broad range of areas such as pharmaceuticals, natural product synthesis or biochemistry.2 Current work in this field focuses on a better understanding of the relationship between the structure and reactivity, application of the reaction to a broader range of substrates (particularly the sterically hindered or electronically deactivated olefins), and alternative operating solvents.3The Hoveyda–Grubbs catalyst is a particularly interesting system, since it allows the introduction of various structural modifications in the benzylidene and isopropyl parts. There are numerous examples of such structural changes and some of these alter the catalytic activities of such compounds to a large degree.4 One notable example was the modification of the isopropoxybenzene part of Hoveyda–Grubbs catalyst into 2,3-dihydrobenzofuran or chromane moiety.5 Authors argued, that the in such complexes the crucial Ru–O distance will be higher than in the parent Hoveyda–Grubbs catalyst resulting in easier Ru–O bond dissociation and faster initiation of the catalyst. The 2,3-dihydrobenzofuran-substituted catalyst has not been, unfortunately, isolated, but the chromane-substituted catalyst showed indeed fast initiation, on the level of the fast Grela nitro catalyst.5,6
The use of natural products and/or biologically-active compounds and ligands as building blocks of new ruthenium-based olefin metathesis catalysts is an interesting approach that provides readily available and well characterized precursors.7 Despite the fact that 2,3-dihydrobenzofuran-substituted Grubb–Hoveyda catalyst has not been isolated, complexes based on the 1-benzofuran core seem to be good candidates as precursors for new olefin metathesis catalysts due to the presence of the O atom, which is able to form a relatively strong Ru–O bond. 1-Benzofurans are often a part of natural products that exhibit activities such as anticancer, antioxidant, antibacterial and antifungal.8 Benzofurans are also found as substructures in a large number of drugs as well as act as the metal chelators and free radical scavengers.9 Due to that facts, there have been a large number of 1-benzofuran analogues synthesized over the course of the last 30 years.
Due to the detailed knowledge of the entire catalytic mechanism and advanced in density functional methods, the search for new generation of metathesis catalysts has, in recent years, moved partially to the computational chemistry field. Cavallo showed recently that Grubbs-type catalysts with Ru replaced by Fe may be a viable option for olefin metathesis.10 Most of these studies focus, however, on new carbene or carbene-like moieties or structural modifications in the isopropyl or benzylidene part of the original Hoveyda–Grubbs catalyst. The most interesting and recent examples of such studies include bis-ylidene Ru complexes,11 nitrenium ions and boron analogues of Arduengo carbenes,12 cyclohexane-modified carbenes obtained using an evolutionary algorithm for de novo ligand optimization,13 and a series of alkoxy-modified chelating benzylidenes.14 These studies show that the computational approach is a feasible option in exploring the vast space of molecular structures when searching for new catalysts to help in their design and synthesis.
We became interested in benzofuran derivatives as Ru-complexes building blocks also due to our preliminary DFT results. We prepared a model (2) of a standard 1st generation Grubbs catalyst,1a,b but with PCy3 replaced by a benzofuran molecule and benzylidene replaced by a methylidene, to avoid any favorable interactions between the benzylidene and benzofuran moieties, as presented in Fig. 1. Our calculations show that benzofuran forms a stable complex with the ruthenium complex and we estimated the Ru–O bond dissociation energy at 17.6 kcal mol−1. This value is very similar to the activation free energy (ΔG‡) of the initiation reaction for Hoveyda–Grubbs catalyst 1 involving the dissociation of the Ru–O bond, estimated experimentally at 19–20 kcal mol−1.13 Due to the similarity of these values we came into the conclusion that benzofuran may be an interesting building block for new class of Ru-based metathesis catalysts.
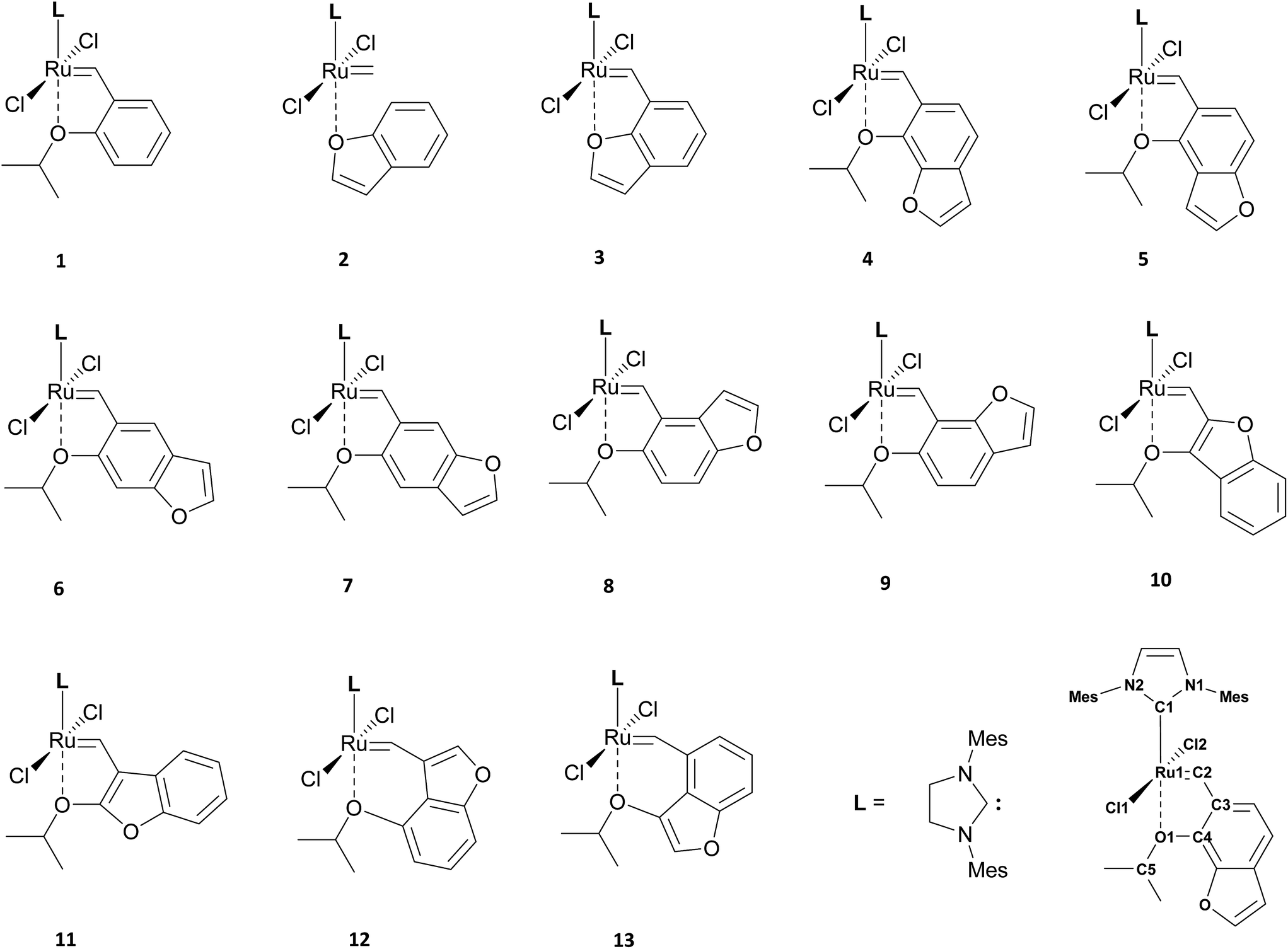 | ||
| Fig. 1 Structure of the candidates for catalysts investigated in this work and numbering scheme used for all complexes. | ||
In this work we performed computational rational design of a new class of ruthenium-based Hoveyda–Grubbs-type catalyst with the o-isopropoxyphenylmethylene part replaced by 1-benzofuran (3) or ten derivatives of isopropoxy-1-benzofuran (4–13, see Fig. 1): (7-isopropoxybenzofuran-6-yl)methylene (4), (4-isopropoxybenzofuran-5-yl)methylene (5), (6-isopropoxybenzofuran-5-yl)methylene (6), (5-isopropoxy benzofuran-6-yl)methylene (7), (5-isopropoxybenzofuran-4-yl)methylene (8), (6-isopropoxybenzofuran-7-yl)methylene (9), (3-isopropoxybenzofuran-2-yl)methylene (10), (2-isopropoxy benzofuran-3-yl)methylene (11), (4-isopropoxybenzofuran-3-yl)methylene (12) and (3-isopropoxybenzofuran-4-yl)methylene (13). We have analysed the impact of these substitutions on the structural and electronic properties of new candidates for catalysts. For all complexes we have also performed calculations estimating the free energy barriers for the metathesis initiation step and compared them with the original Hoveyda–Grubbs catalyst. We also show the relationship between the structure and catalytic activity for this series of compounds, which allowed us to select the best candidates for efficient and stable Ru-based olefin metathesis catalysts, to be synthesized in the future.
Results and discussion
Structural studies
We started our analysis of the new, hypothetical catalysts by finding the energy minima for the isomers with the chlorides in either the cis or trans positions. In the case of 1 it is known that the trans geometry is favored over the cis geometry not only for the precatalyst, but also throughout the entire catalytic reaction.15 On the other hand some similar compounds with a different NHC and other structural modification can adopt the cis conformation.16 Regarding benzofuran-derived systems we found that the trans stereoisomers were favored by 3.3 kcal mol−1 for 3 and between 6.4 and 9.5 kcal mol−1 for all other complexes (see geometry of precatalysts in the ESI†). We also calculated the difference in trans–cis conformation energy for 1, which was found to be 8.5 kcal mol−1. Based on these results and the fact that 1 forms the trans isomer almost exclusively we can claim that for all investigated systems the trans isomer is heavily favored over the cis one and will be solely present in the solution.Table 1 compares the geometrical parameters as well as partial charges (Mulliken, ESP and natural) of all studied derivatives with the original Hoveyda–Grubbs catalyst. As in our and other groups' previous studies there are only small differences in the geometries of these systems.12,14,17 The Ru1–O1 bond for most benzofuran-derived complexes is longer than in the Hoveyda-case, suggesting a weaker Ru1–O1 bond. This is particularly pronounced for system with the five-membered ring of the benzofuran is involved in the formation of Ru–C–C–C–O or Ru–C–C–C–C–O ring – 3, 10, 11, 12 and 13. The longer Ru1–O1 bond shows, however, no correlation with length of the Ru1–C1 bond, which in all cases shows very little variation. The bond dissociation energy (BDE) for the Ru1–C1 bond correlates well (R2 = 0.67) with the Ru1–C1 bond length, what is an expected result. The length of the Ru1–C1 bond does not, however, correlate with the length of the Ru1–C2 bond, which might have been expected due to the trans effect.17,18 Overall, the relatively high rigidity of Hoveyda-type catalysts forbids any major structural changes in systems derived from this complex and, unfortunately, doesn't give any useful information which may be translated into the estimates of their catalytic activity.
| 1 X-ray5 | 1 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Bond length (Å) | |||||||||||||
| Ru1–C2 | 1.829(1) | 1.832 | 1.839 | 1.836 | 1.833 | 1.833 | 1.832 | 1.838 | 1.834 | 1.865 | 1.861 | 1.834 | 1.839 |
| Ru1–C1 | 1.979(1) | 1.969 | 1.949 | 1.966 | 1.968 | 1.968 | 1.969 | 1.970 | 1.968 | 1.950 | 1.941 | 1.968 | 1.975 |
| Ru1–O1 | 2.256(1) | 2.323 | 2.473 | 2.322 | 2.328 | 2.317 | 2.326 | 2.316 | 2.339 | 2.387 | 2.443 | 2.447 | 2.374 |
| O1–C4 | 1.370(2) | 1.352 | 1.351 | 1.345 | 1.349 | 1.351 | 1.356 | 1.355 | 1.335 | 1.335 | 1.307 | 1.357 | 1.346 |
| O1–C5 | 1.469(2) | 1.448 | 1.372 | 1.459 | 1.451 | 1.448 | 1.447 | 1.448 | 1.449 | 1.449 | 1.463 | 1.450 | 1.450 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||||||||||
| Plane angle (degrees) | |||||||||||||
| N1–C1–N2 | 107.20(1) | 106.7 | 107.1 | 106.7 | 106.6 | 106.7 | 106.7 | 106.6 | 106.8 | 106.9 | 106.9 | 106.5 | 106.4 |
| C1–Ru1–C2 | 101.34(6) | 102.4 | 101.7 | 102.6 | 102.5 | 102.8 | 102.0 | 102.8 | 101.7 | 101.8 | 102.3 | 98.7 | 98.1 |
| Cl–Ru1–Cl | 156.25(1) | 160.7 | 158.9 | 161.6 | 160.6 | 161.6 | 160.9 | 160.8 | 160.4 | 160.9 | 160.6 | 163.5 | 162.8 |
|
|||||||||||||
| BDE (kcal mol−1) | |||||||||||||
| Ru1–C1 | 80.86 | 81.41 | 78.62 | 79.26 | 79.07 | 78.36 | 80.29 | 79.10 | 80.78 | 85.20 | 76.74 | 77.27 | |
|
|||||||||||||
| Mulliken partial charges | |||||||||||||
| Ru1 | −0.01 | 0.00 | −0.04 | −0.04 | −0.03 | −0.03 | −0.03 | −0.03 | −0.05 | −0.06 | −0.05 | −0.05 | |
| C1 | 0.36 | 0.44 | 0.44 | 0.44 | 0.44 | 0.44 | 0.44 | 0.44 | 0.44 | 0.44 | 0.45 | 0.44 | |
| O1 | −0.54 | −0.48 | −0.54 | −0.53 | −0.53 | −0.54 | −0.53 | −0.53 | −0.52 | −0.50 | −0.54 | −0.55 | |
| C2 | −0.03 | −0.12 | −0.09 | −0.09 | −0.09 | −0.09 | −0.09 | −0.10 | −0.15 | −0.12 | −0.09 | −0.07 | |
| C4 | 0.39 | 0.29 | 0.39 | 0.42 | 0.40 | 0.40 | 0.39 | 0.38 | 0.40 | 0.64 | 0.47 | 0.41 | |
| C5 | 0.16 | 0.16 | 0.15 | 0.16 | 0.16 | 0.16 | 0.16 | 0.16 | 0.15 | 0.14 | 0.16 | 0.15 | |
|
|||||||||||||
| ESP partial charges | |||||||||||||
| Ru1 | 0.78 | 0.84 | 0.98 | 0.99 | 1.00 | 1.00 | 1.02 | 1.01 | 0.94 | 0.92 | 1.15 | 1.13 | |
| C1 | −0.10 | −0.32 | −0.35 | −0.38 | −0.40 | −0.39 | −0.43 | −0.43 | −0.33 | −0.45 | −0.44 | −0.45 | |
| O1 | −0.20 | −0.38 | −0.26 | −0.27 | −0.36 | −0.33 | −0.33 | −0.39 | −0.25 | −0.38 | −0.48 | −0.40 | |
| C2 | −0.42 | −0.39 | −0.50 | −0.47 | −0.41 | −0.51 | −0.56 | −0.38 | −0.59 | −0.46 | −0.52 | −0.64 | |
| C4 | 0.07 | 0.33 | −0.04 | 0.06 | 0.26 | 0.16 | 0.17 | 0.29 | −0.11 | 0.49 | 0.45 | 0.18 | |
| C5 | 0.17 | 0.09 | 0.32 | 0.27 | 0.32 | 0.33 | 0.32 | 0.35 | 0.31 | 0.33 | 0.38 | 0.35 | |
|
|||||||||||||
| Natural partial charges | |||||||||||||
| Ru1 | 0.38 | 0.32 | 0.30 | 0.30 | 0.31 | 0.31 | 0.31 | 0.31 | 0.31 | 0.28 | 0.32 | 0.32 | |
| C1 | 0.39 | 0.51 | 0.49 | 0.49 | 0.49 | 0.49 | 0.49 | 0.49 | 0.50 | 0.51 | 0.50 | 0.49 | |
| O1 | −0.53 | −0.47 | −0.53 | −0.53 | −0.53 | −0.54 | −0.53 | −0.53 | −0.52 | −0.53 | −0.54 | −0.54 | |
| C2 | 0.06 | −0.01 | 0.01 | 0.01 | 0.01 | 0.00 | 0.00 | 0.00 | −0.07 | 0.00 | 0.01 | 0.02 | |
| C4 | 0.36 | 0.38 | 0.37 | 0.42 | 0.40 | 0.39 | 0.40 | 0.41 | 0.35 | 0.79 | 0.38 | 0.27 | |
| C5 | 0.11 | 0.15 | 0.11 | 0.12 | 0.11 | 0.11 | 0.11 | 0.11 | 0.12 | 0.11 | 0.11 | 0.11 | |
Performance in metathesis initiation
Numerous experimental and computational studies showed that there are three possible initiation paths for the Hoveyda–Grubbs-type catalysts: dissociative, associative and interchange. In the dissociative mechanism the entire catalytic process is initiated by the dissociation of the Ru1–O1 bond, followed by the olefin association. In the associative mechanism the entire catalytic process commences with the olefin association followed by Ru1–O1 bond dissociation. A third possibility is the interchange mechanism, where both events (olefin association and Ru1–O1 bond dissociation) occur simultaneously. The most recent theoretical and experimental results suggest that for all investigated Hoveyda-like and Grubbs-like systems the associative mechanism is always characterized by the highest energy barrier.19 On the other hand the interchange mechanism is often a viable pathway and in many cases and depending on both the catalyst and the olefin substrate, the initiation may follow either the dissociative, interchange or both mechanisms in parallel. In all known cases precatalyst initiation is the rate-limiting step of the olefin metathesis catalytic cycle at low and moderate olefin concentrations.20 Moreover, for large olefins the dissociative path becomes always favorable from the free energy point of view, due to the steric hindrances and the low possibility of an olefin attack on the Ru center before the complete dissociation of the Ru1–O1 bond. As a result, in this study we have only considered the dissociative mechanism as a valid initiation pathways for the new, hypothetical precatalysts.Due to structural differences between the studies complexes we will describe the initiation route for each system separately. Of all investigated cases complex 3 shows the lowest ΔG‡ of only 9.84 kcal mol−1 (see Fig. 2). Such a low value of free energy of activation comes from a relatively weak Ru1–O1 bond with the length of 2.47 Å, which in turn is the consequence of a relatively large strain caused by the benzofuran core. A system with such a weak Ru1–O1 bond is likely to be unstable in solution. This results is consistent with Grela's attempts to synthesize a similar complex (with 1,3-bis(2,4,6-trimethyl-phenyl)imidazol-2-ylidene (IMes) replaced by 1,3-bis(2,4,6-trimethylphenyl)-imidazolin-2-ylidene (SIMes)), who observed no formation of the desired product.5
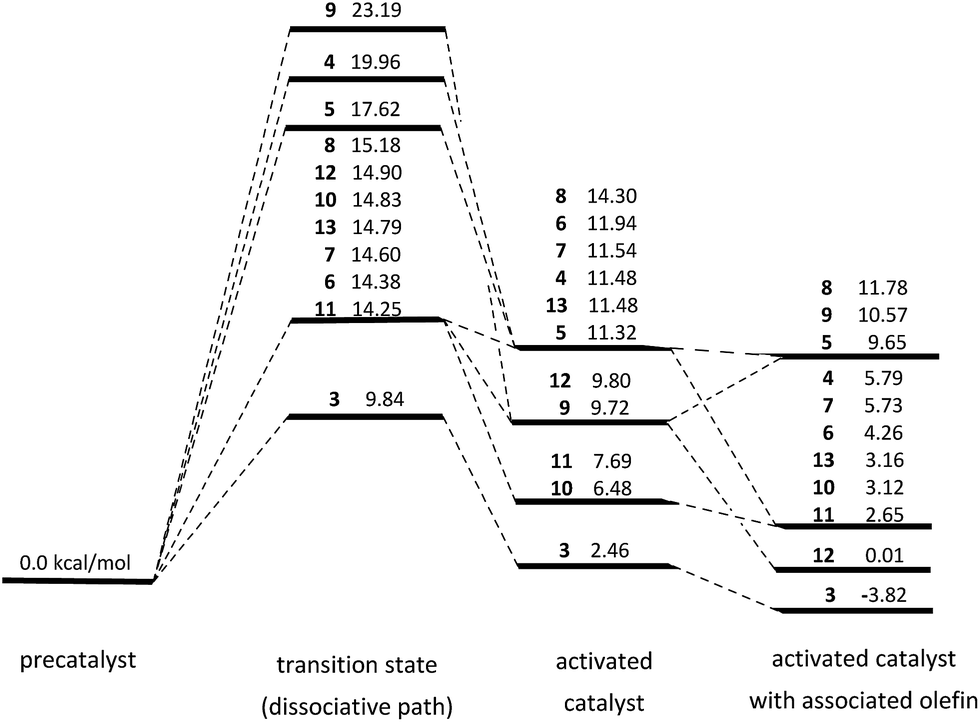 | ||
| Fig. 2 Calculated free energies of activation (ΔG‡) and relative free energies of the precatalyst, activated catalyst and catalyst with the associated olefin of the new Ru-complexes studies in this work. All values are in kcal mol−1 and relative to the corresponding precatalyst. | ||
Complex 4 is, on the other hand, the most similar to a standard Hoveyda–Grubbs catalyst, both from the structural point of view and the free energy profile of the initiation. Structurally, the Ru1–C2–C3–C4–O1 core from 1 is completely preserved and the additional, five-membered ring of the benzofuran is relatively far from Ru ion. As a result the ΔG of activation is estimated at 19.96 kcal mol−1, very similar to the experimental value for 1. Complex 5, for which the coordination mode of the benzofuran moiety is the mirror image of 4, the value of ΔG‡ is slightly lower (17.62 kcal mol−1), while the geometries of the precatalyst, transition state and activated catalyst are very similar. These two complexes also have all major geometrical parameters almost identical to the Hoveyda–Grubbs catalyst.
Complexes 6 and 7 form another similar pair, with the only difference between them being the position of the O atom. Due to such a small difference in their structure we expected similar results for this pair, and this is indeed the case: ΔG‡ for 6 is estimated at 14.38 kcal mol−1, while for 7 we obtained the value of 14.60 kcal mol−1. These values are, however, clearly lower than in the case of the 4/5 pair and the original Hoveyda–Grubbs catalyst 1, suggesting a much faster initiation. We can explain the difference between these pairs on the basis of aromatic stabilization of the precatalysts, similarly to an older work in which a series of naphthalene-substituted Hoveyda-type systems was synthesized and characterized.21 In this work a series of three new complexes, depicted in Fig. 3, has been synthesized and tested for catalytic activity in model ring-closing metathesis (RCM) reactions. Interestingly, two catalysts (14 and 16) showed a close to none activity in RCM reactions, while complex 15 showed moderate activity, on the level of Hoveyda–Grubbs catalyst. Authors of this work noticed that the structure of 15 resembles anthracene, if we consider the Ru1–C22–C23–C28–O1 substructure as the third aromatic ring, while in 14 and 16 a structure similar to phenantrene is formed. Using this simple approach and taking the nucleus-independent chemical shift (NICS) values for outer rings of anthracene (−8.2) and phenantrene (−10.2)22 we can explain the high inactivity of 14 and 16 via high aromatic stabilization of the Ru1–C22–C23–C28–O1 ring. On the other hand the anthracene-like complex 15 behaves similarly to naphthalene-like Hoveyda–Grubbs catalyst, giving similar activities and, likely, similar values of ΔG‡ of initiation.
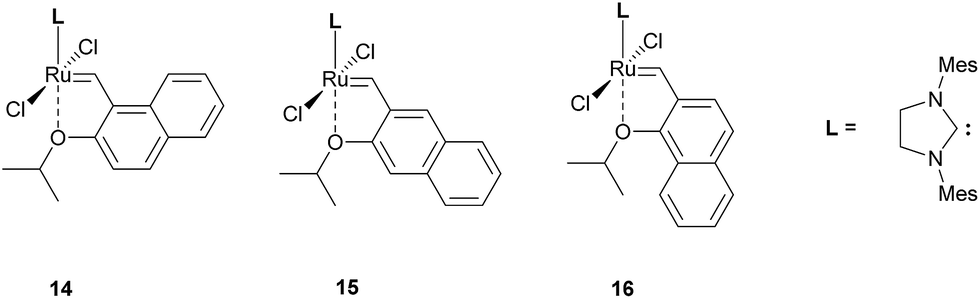 | ||
| Fig. 3 Hoveyda–Grubbs-type ruthenium complexes with the benzylidene part replaced by vinylnaphthalene (adopted from ref. 21). | ||
In the case of our systems and 6/7 versus 4/5 difference we can apply a similar reasoning, albeit taking into the account the presence of the heteroatom in the benzofuran core. Interestingly, benzofuran has very similar aromatic properties to naphthalene, resulting in very similar NICS values for the six-membered ring (−9.6) and the five-membered ring (−9.3).23 As a result we should expect similar chemical properties between ruthenium complexes based on benzofuran core and can split them into two groups. The first group, structural analogues of 14 (8 and 9) and 16 (4 and 5) should form relatively inactive catalysts with a relatively high ΔG‡, while the second group, structural analogues of 15 (6 and 7) should form relatively active catalysts with a relatively low ΔG‡. As we have already seen this is true for 4 and 5, though our computational results suggest that these catalysts possess similar activity in metathesis as Hoveyda–Grubbs catalyst 1 and do not become almost completely inactive as complexes 14 and 16. The likely cause of such behavior is the presence of both the five-membered ring and the O heteroatom, which make the Ru1–C22–C23–C28–O1 ring in 4 and 5 less aromatic. This is also true for 6 and 7, which show much lower ΔG‡ values, although our computational results suggest they should be actually much faster than Hoveyda–Grubbs catalyst 1.
For the third pair consisting of 8 and 9 complexes, analogues of 14 with phenantrene-like structure we would expect therefore a relatively large ΔG‡ values and relatively low activity. Our results are, however, inconsistent with these expectations. First, ΔG of activation of 8 is 15.18 kcal mol−1, less than 1 kcal mol−1 more than for 6/7 pair and well below ΔG‡ values for 4/5. There is no simple explanation of this phenomenon on the basis of the NICS/aromacity concept. It is, though, worth noting that the difference in free energy between the activated 8 complex and the free energy of the initiation transition state is below 1 kcal mol−1 (see Fig. 2), which is by far the smallest difference for all studied candidates for catalyst. As a result, there is a very high probability of deactivation of the catalyst and reverting back to the precatalyst, instead of coordinating the olefin and starting the catalytic cycle. Additionally, upon the coordination of the olefin the free energy of the system remains relatively large with respect to both the precatalyst and the activated catalyst, reinforcing the hypothesis of 8 being a poor candidate for an efficient catalyst.
On the other hand complex 9 is characterized by a large ΔG‡ of activation (23.19 kcal mol−1) and a relatively stable activated catalyst. A look at the geometry of the precatalyst and activated catalyst reveals, however, that there is a Ru–O interaction in both of these conformations (see Fig. 4), though formed by different oxygen atoms of the benzofuran core. Consequently, the “activation” of 9 via Ru1–O1 bond dissociation leads to the formation of a different Ru1–O bond, rendering the “activated” form inactive and disallowing the association of the olefin. The ΔG value for the activated catalyst with associated olefin, given for 9 in Fig. 2 corresponds to a local minimum geometry with the Ru1–C(olefin) distance of more than 4 Å and virtually no coordination of the olefin to the Ru center.
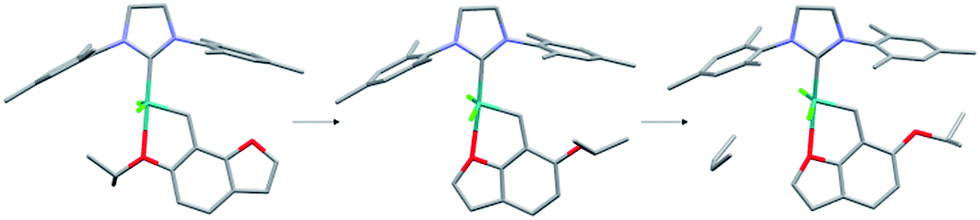 | ||
| Fig. 4 Activation path for complex 9 resulting in formation of a different Ru–O bond and no possibility of olefin association. | ||
The last four investigated complexes represent a group in which the metalloaromatic ring is formed partially by the five-membered ring of benzofuran. In the case of complexes 10 and 11 the ring is composed of Ru1 and O1 atoms and three carbon atoms, similarly to 1 and 4–9. The difference lies in the length of the C23–C28 bond of the five-membered ring and different C22–C23–C28 and C23–C28–O1 angles which the Ru1–O1 bond length. The shorter bond (1.38 Å for both 10 and 11 versus 1.41–1.43 Å for complexes 4–9) and larger sum of C22–C23–C28 and C23–C28–O1 angle values (240.8° for 10 and 242.5° for 11 versus 232.9° for 1) forces the O1 atom away from Ru1 with respect to the Hoveyda–Grubbs catalyst. The final result is consistent with this analysis, as the Ru1–O1 bond becomes longer and weaker than in 1, leading to lower values of ΔG of activation.
Finally, complexes 12 and 13 form a pair with a completely different bonding scheme, with the metalloaromatic ring formed by Ru1, O1 and four carbon atoms (instead of three as in all other cases). Similarly to the previous pair such an arrangement forces the O1 atom away from Ru1 with respect to 1, making the Ru1–O1 bond longer and the values of ΔG‡ of initiation smaller. In analogy to 12/13, this pair seems to consist of good candidates for new Ru-based metathesis catalysts.
Conclusions
There is a wealth of theoretical investigations on ruthenium-based metathesis catalysts and they show that modern computational DFT methods are able to recreate most experimental findings (structural data, initiation rates, free energies, enthalpies and entropies of activation) for standard olefin metathesis catalysts with good accuracy. Due to that, computational studies are becoming more and more helpful in guiding synthetic chemists in further preparation of new catalysts as well as contributing to a better understanding of the structure–activity relationships for this class of compounds. In the particular case of Ru-based metathesis catalysts there is also a constant search for correlation between different structural parameters as well as many different parameters obtained with various experimental and computational techniques for the precatalyst and the initiation rate and/or free energy of activation.12 If found, such a correlation would provide a fast and possibly cost-effective method of estimating the catalytic activity of a given complex without performing the actual catalytic reactions, or (in the case of computational predictions) without synthesizing the complex.The most important and reliable method for computational prediction of the catalytic activity is the estimation of the free energy barrier for the slowest, rate-determining step of the catalytic cycle. In the case of Hoveyda–Grubbs-like complexes this step is, in most cases, the initial activation of the precatalyst and it has been shown that computational estimations of ΔG of activation agree quantitatively with the experimental values of both ΔG‡ and initiation rates for a broad range of species in this class of complexes.12,24 In this work we have used the same approach to estimate ΔG‡ for a series of Hoveyda–Grubbs-like systems with the isopropoxybenzene part replaced by 1-benzofuran or ten isopropoxy-1-benzofuran derivatives. Eight of the newly proposed complexes (5–8, 10–13) show ΔG‡ values 2–5 kcal mol−1 lower than the Hoveyda–Grubbs catalyst, which should translate to approximately 10–1000 times faster initiation at 25 °C, resulting in efficient and fast catalysts on the level of Grela nitro catalyst.6 For one of the investigated complex (4) the estimates of ΔG‡ shows value nearly identical with the Hoveyda–Grubbs catalyst. Finally, 3 and 9 are clearly poor candidates for catalysts – one due to high instability and the other due to structural features, which prevent the olefin association to the Ru ion. We hope that these results will encourage experimental groups to synthesize complexes 5–8, 10–13, which can be obtained from appropriate precursors (listed in ESI†) to experimentally test their catalytic activities.
We additionally searched for correlations between the obtained ΔG‡ values and computed structural or other parameters, including partial charges computed using three different approaches. The best correlation, albeit still rather weak (R2 = 0.32), was found between the ΔG‡ values and Ru1–O1 bond lengths of the precatalyst. Since the initiation of the precatalyst relies on the Ru1–O1 bond breaking, which is easier if the bond is longer, it is a rather obvious result. No other meaningful correlation was found in this study suggesting, that the only reliable measure of the catalytic activity is, for now the estimation of the ΔG‡, a relatively difficult and time-consuming process due to the necessity of locating one or more transitions states involved in the precatalyst activation.
Experimental
Computational details
In this study we used density functional theory (DFT) calculations to study the structures of investigated complexes and possible pathways of their initiation mechanisms. The calculations have been performed using a computational protocol similar to our previous studies.12,17,25 We have used an all-atom model for all studied catalysts and the cis-2-butene molecule to model the substrate of olefin metathesis. Starting models for precatalyst were prepared on the basis of available CSD crystal structures of a well-known Hoveyda precatalyst (refcode: ABEJUM01).5 In the first step, all structures were modeled using the M06-D3 density functional with the 6-31G** basis set for all atoms except the Ru atom, which was described by the Los Alamos angular momentum projected effective core potential (ECP) using the double-ζ contraction of valence functions (denoted as LACVP**).26 We have chosen M06 functional, since it was also shown to perform particularly well for ruthenium-based catalysts, giving accurate energies for a number of Grubbs and Hoveyda systems.27 Since the M06 functional has already medium-range dispersion implemented, M06-D3 may overestimate the effect of dispersion due to double-counting of these effects.28 On the other hand the addition of D3 correction to M06 was shown to improve the results for many organic reactions when calculating the differences in relative energies.26We have used the standard energy convergence criterion of 5 × 10−5 Hartree. For each structure frequencies were calculated to verify the nature of each stationary point (zero imaginary frequencies for minima and one imaginary frequency for transition states). In the second step we calculated solvation energies using the Poisson–Boltzmann self-consistent polarizable continuum method (PBF) as implemented in Jaguar v.7.9 (Schrodinger, 2013) to represent dichloromethane, using the dielectric constant of 8.93 and the effective radius 2.33 Å. The solvation calculations were performed using the M06-D3/LACVP** level of theory and the gas-phase optimized structures. For all stationary points we have also performed single-point energy calculations using the same M06 functional, but with a larger basis set: here Ru was described with the triple-ζ contraction of valence functions augmented with two f functions, and the core electrons were described by the same ECP; the other atoms were described with the 6-311++G** basis set.
Energies discussed in this work for stationary points are free energies, calculated as the sum of electronic energy (single-point, using the larger 6-311++G** basis set), solvation energy, zero-point energy correction, thermal correction to enthalpy, and the negative product of temperature and entropy (at 298 K). For calculating bond dissociation energies we used the same 6-311++G** basis set and counterpoise correction using the standard Boys–Bernardi scheme.29
Acknowledgements
This work was supported by NCN grant. UMO-2012/05/B/ST5/00715. The calculations were partially performed at the Interdisciplinary Center for Mathematical and Computational Modelling (University of Warsaw) under G53-9 computational grant.Notes and references
- (a) M. Scholl, T. M. Trnka, J. P. Morgan and R. H. Grubbs, Tetrahedron Lett., 1999, 40, 2247 CrossRef CAS; (b) J. Huang, E. D. Stevens, S. P. Nolan and J. L. Petersen, J. Am. Chem. Soc., 1999, 121, 2674 CrossRef CAS; (c) J. Huang, H.-J. Schanz, E. D. Stevens and S. P. Nolan, Organometallics, 1999, 18, 5375 CrossRef CAS; (d) S. B. Garber, J. S. Kingsbury, B. L. Gray and A. H. Hoveyda, J. Am. Chem. Soc., 2000, 122, 8168 CrossRef CAS.
- (a) G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746 CrossRef CAS PubMed; (b) C. Samojłowicz, M. Bieniek and K. Grela, Chem. Rev., 2009, 109, 3708 CrossRef PubMed.
- (a) M. J. Koh, R. K. M. Khan, S. Torker, M. Yu, M. S. Mikus and A. H. Hoveyda, Nature, 2015, 517, 181 CrossRef CAS PubMed; (b) J. S. Cannon and R. H. Grubbs, Angew. Chem., Int. Ed., 2013, 52, 9001 CrossRef CAS PubMed; (c) K. Skowerski, J. Białecki, A. Tracz and T. K. Oleszewski, Green Chem., 2014, 16, 1125 RSC; (d) Ł. Gulajski, P. Śledź, A. Lupa and K. Grela, Green Chem., 2008, 10, 279 RSC; (e) B. H. Lipshultz, G. T. Aguinaldo, S. Ghorai and K. Voigritter, Org. Lett., 2008, 7–8, 953 Search PubMed; (f) S. J. Connon and S. Blechert, Bioorg. Med. Chem. Lett., 2007, 12, 1873–1876 CrossRef.
- (a) Y. Vidavsky, A. Anaby and N. G. Lemcoff, Dalton Trans., 2012, 41, 32 RSC; (b) K. M. Engle, G. Lu, S.-X. Luo, L. M. Henling, M. K. Takase, P. Liu, K. N. Houk and R. H. Grubbs, J. Am. Chem. Soc., 2015, 137, 5782 CrossRef CAS PubMed.
- M. Barbasiewicz, M. Bieniek, A. Michrowska, A. Szadkowska, A. Makal, K. Woźniak and K. Grela, Adv. Synth. Catal., 2007, 349, 193 CrossRef CAS.
- A. Michrowska, R. Bujok, S. Harutyunyan, V. Sashuk, G. Dolgonos and K. Grela, J. Am. Chem. Soc., 2004, 126, 9318 CrossRef CAS PubMed.
- (a) E. C. Gleeson, Z. J. Wang, W. R. Jackson and A. J. Robinson, J. Org. Chem., 2015, 80, 7205 CrossRef CAS PubMed; (b) A. Hryniewiecka, J. W. Morzycki and S. Witkowski, J. Organomet. Chem., 2010, 695, 1265 CrossRef; (c) A. Hryniewiecka, J. W. Morzycki, L. Siergiejczyk, S. Witkowski, J. Wójcik and A. Gryf-Keller, Aust. J. Chem., 2008, 62, 1363 CrossRef; (d) A. Hryniewiecka, A. Kozłowska and S. Witkowski, J. Organomet. Chem., 2012, 701, 87 CrossRef.
- (a) H. K. Shamsuzzaman, Eur. J. Med. Chem., 2015, 97, 483 CrossRef PubMed; (b) J. Kossakowski, K. Ostrowska, M. Struga and J. Stefańska, Med. Chem. Res., 2009, 18, 555 CrossRef CAS; (c) E. Hejchman, K. Ostrowska, D. Maciejewska, J. Kossakowski and W. E. Courchesne, J. Pharmacol. Exp. Ther., 2012, 343, 380 CrossRef CAS PubMed; (d) K. Ostrowska, E. Hejchman, I. Wolska, H. Kruszewska and D. Maciejewska, Monatsh. Chem., 2013, 144, 1679 CrossRef CAS PubMed.
- (a) W. E. Courchesne, E. Hejchman, D. Maciejewska, J. Kossakowski and K. Ostrowska, Antifungal compounds, US Pat., App. 13/330,349, 2011; (b) T. Pena Ruiz, A. Drzewiecka, A. E. Koziol, M. F. Gomez, K. Ostrowska, M. Struga and J. Kossakowski, Struct. Chem., 2012, 23, 1563 CrossRef; (c) T. Pena Ruiz, A. Drzewiecka, A. E. Koziol, M. F. Gomez, K. Ostrowska, M. Struga and J. Kossakowski, Struct. Chem., 2012, 23, 1617 CrossRef; (d) A. Drzewiecka, A. E. Koziol, M. T. Klepka, A. Wolska, S. B. Jimenez-Pulido, T. Lis, K. Ostrowska and M. Struga, Chem. Phys. Lett., 2013, 559, 41 CrossRef CAS.
- (a) A. Poater, E. Pump, S. V. C. Vumaletti and L. Cavallo, Chem. Phys. Lett., 2014, 610–611, 29 CrossRef CAS; (b) A. Poater, S. V. C. Vumaletti, E. Pump and L. Cavallo, Dalton Trans., 2014, 43, 11216 RSC.
- A. Poater, R. Credendino, C. Slugovc and L. Cavallo, Dalton Trans., 2013, 42, 7271 RSC.
- A. Pazio, K. Woźniak, K. Grela and B. Trzaskowski, Dalton Trans., 2015, 44, 20021 RSC.
- Y. Chu, W. Heyndrickx, G. Occhipinti, V. R. Jensen and B. K. Alsberg, J. Am. Chem. Soc., 2012, 134, 8885 CrossRef CAS PubMed.
- K. M. Engle, G. Lu, S.-X. Luo, L. M. Henling, M. K. Takase, P. Liu, K. N. Houk and R. H. Grubbs, J. Am. Chem. Soc., 2015, 137, 5782 CrossRef CAS PubMed.
- D. Benitez, E. Tkatchouk and W. A. Goddard, Chem. Commun., 2008, 6194 RSC.
- (a) E. Pump, A. Poater, M. Zirngast, A. Torvisco, R. Fischer, L. Cavallo and C. Slugovc, Organometallics, 2014, 33, 2806 CrossRef CAS; (b) C. E. Diesendruck, E. Tzur, A. Ben-Asuly, I. Goldberg, B. F. Strau and N. G. Lemcoff, Inorg. Chem., 2009, 48, 10819 CrossRef CAS PubMed.
- A. Pazio, K. Woźniak, K. Grela and B. Trzaskowski, Organometallics, 2015, 34, 563 CrossRef CAS.
- H.-C. Yang, Y.-C. Huang, Y.-K. Lan, T.-Y. Luh, Y. Zhao and D. G. Truhlar, Organometallics, 2011, 30, 4196 CrossRef CAS.
- I. W. Ashworth, I. H. Hillier, D. J. Nelson, J. M. Percy and M. A. Vincent, Chem. Commun., 2011, 47, 5428 RSC.
- (a) V. Thiel, M. Hendann, K.-J. Wannowius and H. J. Plenio, J. Am. Chem. Soc., 2012, 134, 1104 CrossRef CAS PubMed; (b) T. Vorfalt, K.-J. Wannowius and H. J. Plenio, Angew. Chem., 2010, 122, 5665 CrossRef.
- M. Barbasiewicz, A. Szadkowska, A. Makal, K. Woźniak and K. grela, Chem.–Eur. J., 2008, 14, 9330 CrossRef CAS PubMed.
- T. M. Krygowski and M. K. Cyrański, Chem. Rev., 2001, 101, 1385 CrossRef CAS PubMed.
- A. Martínez, M.-V. Vázquez, J. L. Carreón-Macedo, L. E. Sansores and R. Salcedo, Tetrahedron, 2003, 59, 6415 CrossRef.
- (a) C. A. Urbina-Blanco, A. Poater, T. Lebl, S. Manzini, A. M. Z. Slawin, L. Cavalli and S. P. Nolan, J. Am. Chem. Soc., 2013, 135, 7073 CrossRef CAS PubMed; (b) A. Poater, E. Pump, S. V. C. Vummaleti and L. Cavallo, J. Chem. Theory Comput., 2014, 10, 4442 CrossRef CAS PubMed; (c) Y. Minenkov, G. Occhipinti and V. R. Jensen, Organometallics, 2013, 32, 2099 CrossRef CAS.
- (a) K. Żukowska, A. Szadkowska, B. Trzaskowski, A. Pazio, Ł. Pączek, K. Woźniak and K. Grela, Organometallics, 2013, 32, 2192 CrossRef; (b) B. Trzaskowski and K. Grela, Organometallics, 2013, 32, 3625 CrossRef CAS.
- S. Luo, Y. Zhao and D. G. Truhlar, Phys. Chem. Chem. Phys., 2011, 13, 13683 RSC.
- (a) D. Benitez, E. Tkatchouk and W. A. Goddard, Chem. Commun., 2008, 6194 RSC; (b) D. Benitez, E. Tkatchouk and W. A. Goddard, Organometallics, 2008, 28, 2643 CrossRef.
- L. Goerikg, J. Phys. Chem. Lett., 2015, 6, 3891 CrossRef PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Geometric parameters, energies and Cartesian coordinates of all investigated systems. See DOI: 10.1039/c6ra01194b |
| This journal is © The Royal Society of Chemistry 2016 |