DOI:
10.1039/C6RA00589F
(Paper)
RSC Adv., 2016,
6, 30661-30682
Exploration of 6,7-dimethoxyquinazoline derivatives as dual acting α1- and AT1-receptor antagonists: synthesis, evaluation, pharmacophore & 3D-QSAR modeling and receptor docking studies†
Received
8th January 2016
, Accepted 15th March 2016
First published on 17th March 2016
Abstract
The 6,7-dimethoxyquinazoline scaffold was further explored to provide dual acting α1- and AT1-receptor antagonists by synthesizing a series of derivatives and biologically evaluating the newly synthesized compounds. Based on the biological data of the current compounds and the earlier reported compounds, pharmacophore models were developed for α1- and AT1-receptor antagonist activities. Subsequently, 3D-QSAR models were also derived for antagonism for both the receptors. The developed 3D-QSAR models were validated using various statistical parameters and both the developed models were further validated using terazosin and prazosin as external compounds. Docking studies confirmed receptor–ligand stabilizing interactions of the balanced-dual active antagonist (110) in the active sites of both α1- as well as AT1-receptors, the structures of which were obtained by homology modeling. Two (42 and 110) of the compounds from the newly synthesized derivatives offered the highest potency (pA2 for α1 = 9.45 and 8.77 and AT1 = 8.36 and 8.60 respectively) with balanced modulation of both the receptors. Both the compounds were found to be slightly less potent to terazosin as α1-antagonists and equipotent to losartan as AT1-antagonists in the in vivo animal model.
1. Introduction
About 17 million deaths are caused globally by cardiovascular complications, out of which about 9.4 million people die due to hypertensive disorders.1 Due to the complexity involved in the pathogenesis of hypertension and affiliated cardiovascular disorders, it is somewhat tricky to manage these disorders. An effective antihypertensive medication is required to fulfill the notable medical deprivation over a broad range of patients as monotherapy.2 Most of the patients with elevated blood pressure (BP) require a combination of two or more drugs to achieve the desired control in BP.3 Therapeutically used drugs basically cause diuresis, adrenoceptor embarrassment or vasodilation. Five categories of drugs such as diuretics, adrenergic receptor blockers, calcium channel blockers, angiotensin converting enzyme (ACE) inhibitors and AT1 (subtype of angiotensin II) receptor antagonists are employed as therapeutically effective antihypertensive agents. There is no single drug or drug category capable of effectively controlling the elevated BP. Many drug combinations like calcium channel blocker (amlodipine) + ACE antagonist (benazepril), diuretic (hydrochlorothiazide) + ACE antagonist (enalapril) etc. are used in the management of hypertension.4,5
To achieve predictable pharmacokinetic and pharmacodynamic correlation and high patient compliance a need was felt for the development of compounds having the ability to act simultaneously at multiple sites.6 Very few compounds have been designed as multiple targeted ligands for the projected targets. Rational designing aspects involve fusion of structural attributes of some selected molecules into a new single entity that could regulate the desired targets.7,8 Balanced activities at the projected targets with high selectivity and satisfactory pharmacokinetics profile are the crucial challenges to design such multitargeted ligands.9
In the designing of multitargeted ligands, the pharmacophores of two or more drugs may be joined together through a cleavable/noncleavable linker or by overlapping of the pharmacophores with each other in a single chemical entity.10 Some publications have appeared11,12 on the designing and development of multiple targeted molecules which are of interest in this field. There are examples of hybrid molecules capable of binding simultaneously to two or more receptors, such as L-746072,13 BMS-346567 (ref. 14) and BMS-248360 (ref. 15) as dual acting receptor antagonists which act on AT1 and ETA (endothelin-A) receptors.16,17
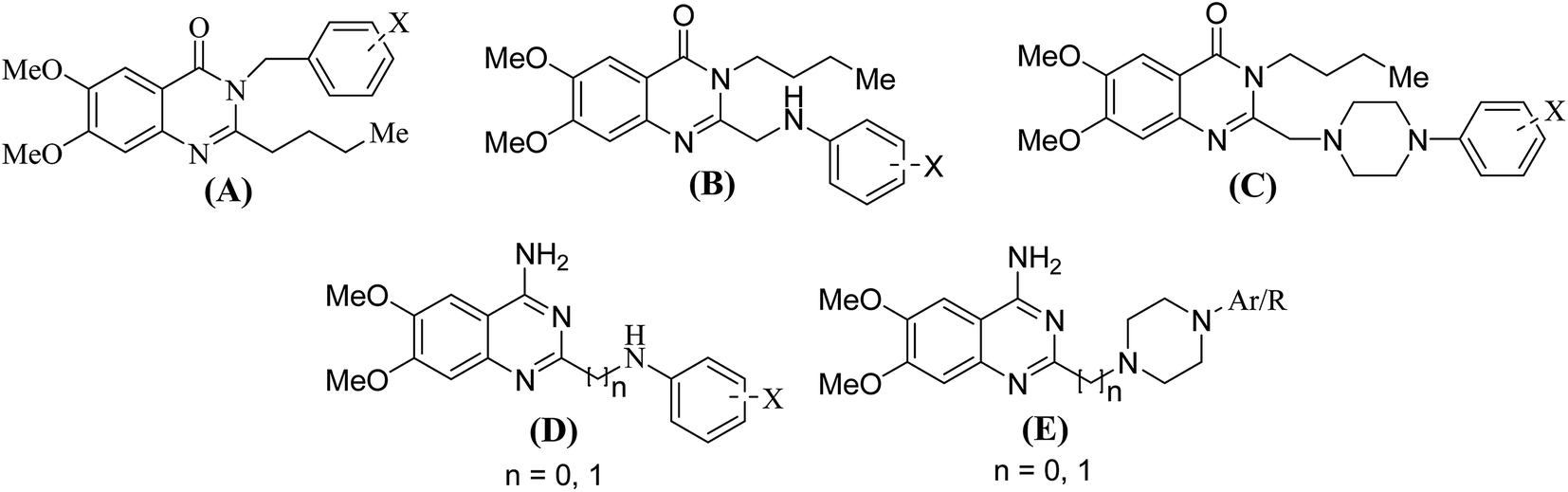
Sympathetic nervous system (SNS) and rennin angiotensin aldosterone system (RAAS) have a balancing effect in regulation of cardiovascular activities17,18 due to cross talks with each other. Due to the dynamic nature of the cross talks, modulation of one system affects the working of the other one. In this situation, simultaneous suppression of SNS and RAAS can have a profound effect on the BP regulation mechanism. Considering the above facts, we designed and reported19 the following five (A–E) categories of 6,7-dimethoxyquinazoline derivatives.
It was tried to incorporate pharmacophoric features of prazosin (1) a prototype of α1-receptor antagonists and AT1-receptor antagonists into a single chemical entity, to obtain dual (α1- and AT1-) antagonists. Initial results of the biological screening of these compounds19 were somewhat surprising as some of the compounds bearing neutral or basic side chains showed very good AT1-antagonistic activity while some others having acidic group showed good α1-antagonistic activity, contrary to the existing assumptions. One such compound (3) exhibited higher activities on α1- as well as AT1-receptors in comparison to the standard drugs prazosin (1) and losartan (2) in the isolated tissue experiments.
It was observed in our earlier study that compounds of type (E) bearing a one carbon spacer between the quinazoline and the piperazine rings offered more balanced and potent dual antagonist activity than compounds without a methylene spacer. To see the effect on biological activity of further increasing the distance between the piperazine ring and the aromatic ring, it was decided to introduce one/two carbon spacers between the two groups (type F). Since a methylene bridge between the quinazoline and the piperazine rings offered superior compounds, it was also planned to have a bare heterocyclic ring system attached to the methylene bridge (G). Additionally, more number of compounds of type (A–E) were also planned to be synthesized to see the impact of different types of groups on the biological activity. It was also aimed to develop a pharmacophore model and validate it by 3D-QSAR model for α1- and AT1-antagonistic activities for these compounds. Therefore, it became imperative to synthesize more number of compounds for each series.
2. Result and discussion
2.1. Chemistry
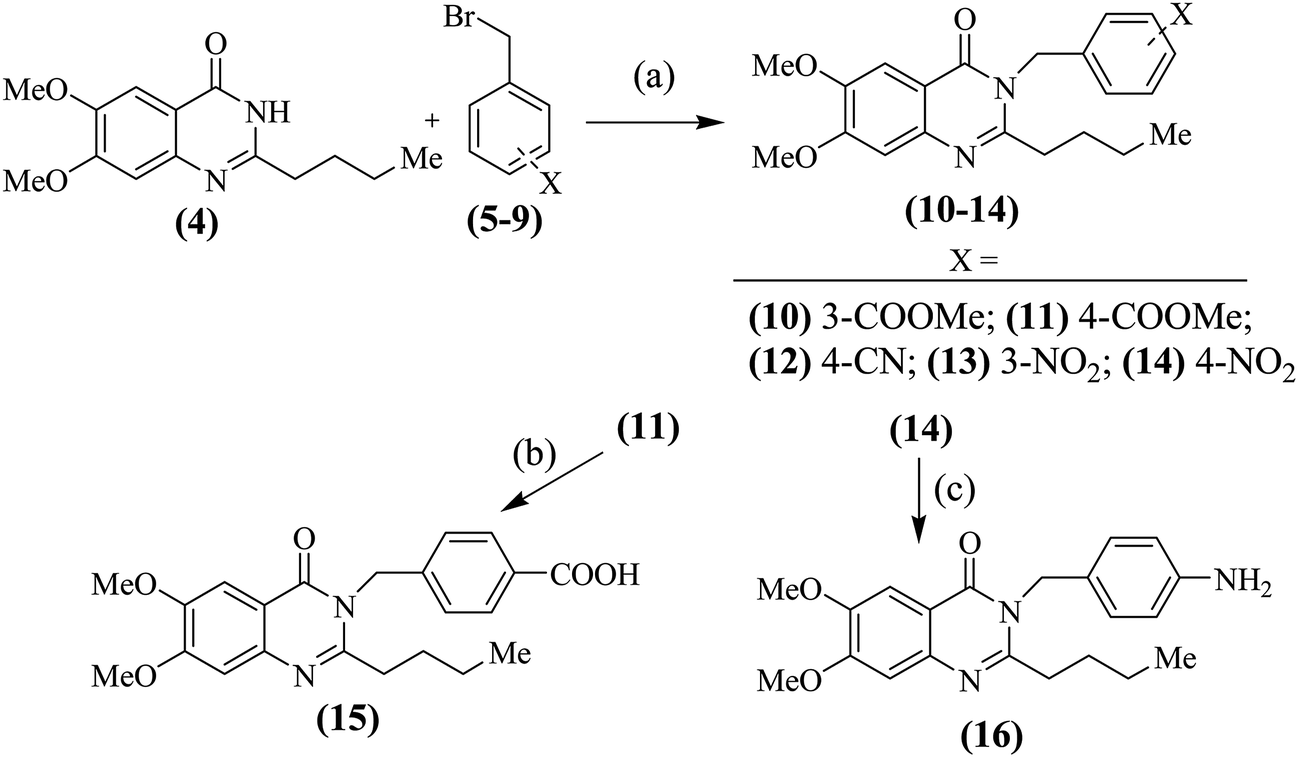
For the synthesis of compounds of types (A–E), the earlier described19 schemes were followed. 2-Butylquinazolinones (10–16) were prepared as per Scheme 1. 2-Butylquinazolinone (4) was reacted with substituted benzyl bromides (5–9) to obtain the N-substituted derivatives (10–14). The 4-methyl ester (11) was hydrolyzed to the free acid (15) in alkaline conditions while, the 4-nitro derivative (14) was reduced to the free amine (16) in presence of iron powder and catalytic amounts of sodium chloride in methanolic solution (Scheme 1).
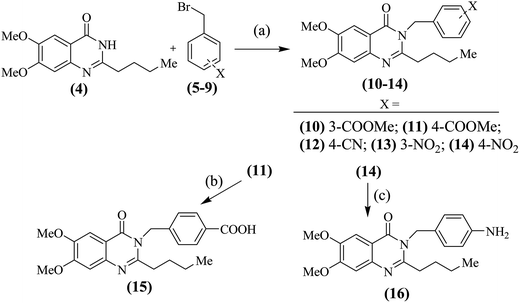 |
| | Scheme 1 Synthesis of compounds (10–16); (a): anh. K2CO3 in dry acetone; (b): aq. KOH (10%) in methanol; (c): iron powder in methanol. | |
3-Butylquinazolinones of types (B and C) were prepared as per Scheme 2. The required 2-chloromethyl derivative19 (17) was reacted with the respective anilines (18–23) to obtain the anilinomethyl derivatives (24–29). In another reaction, the 2-chloromethyl derivative19 (17) was reacted with N-substituted piperazines (30–32) to obtain the desired piperazinomethyl derivatives (33–35).
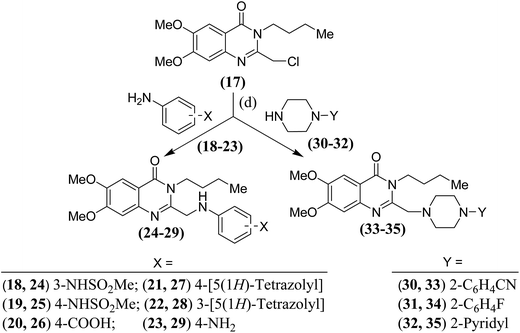 |
| | Scheme 2 Synthesis of compounds (24–29 and 33–35); (d): anh. CsCO3/K2CO3 in dry DMF. | |
In the 4-aminoquinazoline series (D & E; n = 0), direct attachment of the nitrogen functionality was achieved by reacting the 2-chloro derivative19 (36) either with the substituted piperazines (39–41) to obtain the 2-piperazino derivatives (42–44) or with m-phenylenediamine (37) to get the 2-(3-anilino) derivative (38) as per Scheme 3.
 |
| | Scheme 3 Synthesis of compounds (38, 42–44); (d): anh. K2CO3 in dry DMF. | |
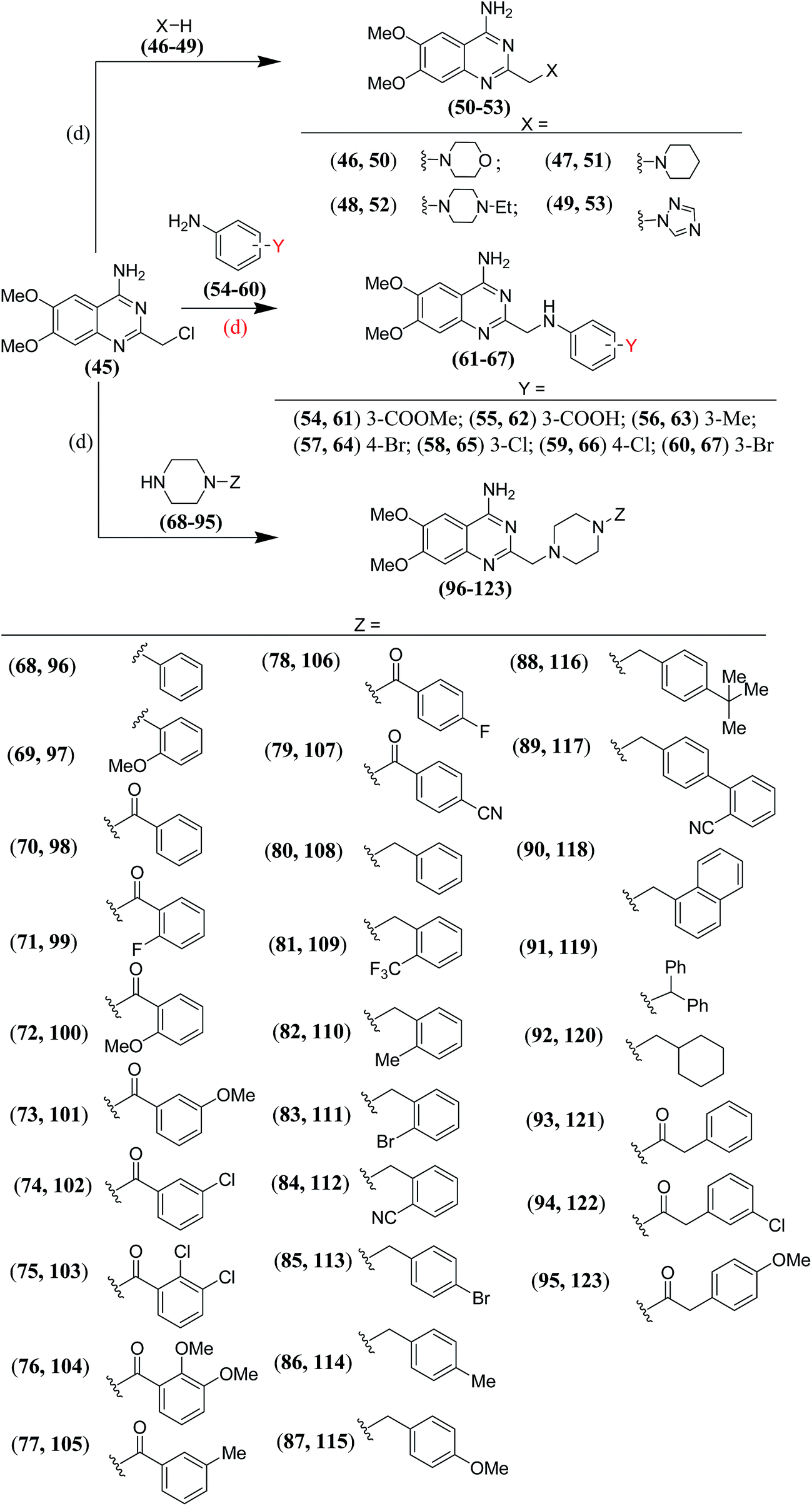
For the synthesis of amino derivatives (50–53) with one carbon spacer between the quinazoline ring and the heterocycle (type G), the 2-chloromethyl derivative19 (45) was reacted directly with various amines (46–49) to obtain the desired derivatives (Scheme 4). Reaction of the 2-chloromethyl derivative19 (45) with substituted anilines (54–60) offered the desired anilinomethyl derivatives (61–67) (type D; n = 1) (Scheme 4).
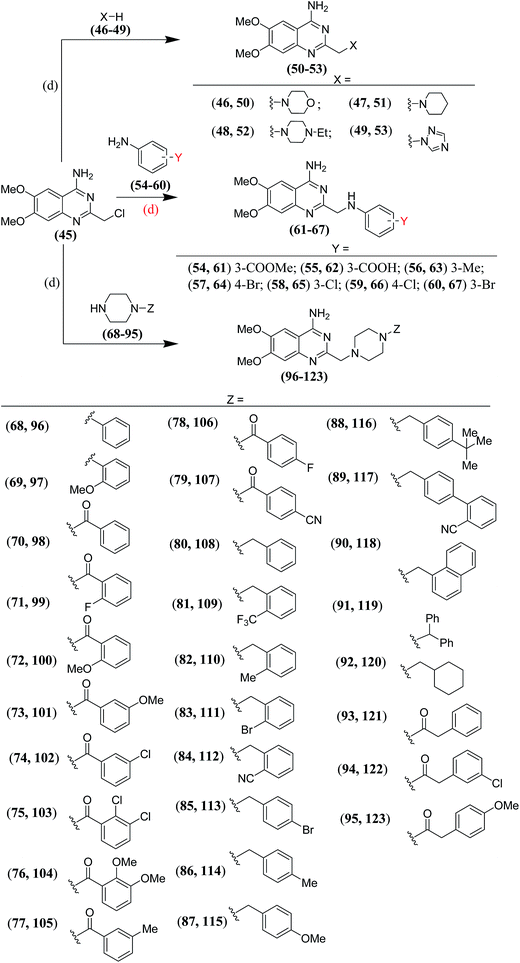 |
| | Scheme 4 Synthesis of compounds (50–53, 61–67 and 96–123); (d): anh. K2CO3 in dry DMF. | |
In our earlier work19 we observed that compounds having 4-aminoquinazoline ring separated by a methylene spacer to the N-arylpiperazine moiety showed potent and balanced α1/AT1 dual antagonistic activity. In these compounds the aryl moiety (Z) was directly linked to the piperazine ring.20–27 So, it was envisaged to introduce one/two carbon spacer between the piperazine ring and the aryl moiety, and observe the impact of this change on the α1/AT1 dual antagonistic activity. The spacer was introduced either as a keto group to ward off the basicity of the attached piperazine nitrogen or as a methylene group to allow it to retain its basic character. In some other compounds two carbon linker was also introduced in place of a keto/methylene linker. To change the physicochemical characteristics of the resulting compounds (96–123), substituents (Z) with different electronegativities and sizes were attached to the linker (Scheme 4).
2.2. In vitro functional antagonism assay
The synthesized compounds were screened for their antagonistic activity on both α1- and AT1-receptors separately by in vitro screening method (Table 1). Compounds of type (A, B and C) having lipophilic (n-butyl) side chain either at the 2nd or 3rd position of 6,7-dimethoxyquinazolin-4-one ring showed moderate (pA2 4–7) antagonistic activity on both (α1- and AT1-) the receptors. Among these, compound (33) having a cyano group at 2nd position on phenyl ring was found to exhibit balanced activity on both the receptors (pA2 7.71 for α1- and 7.41 for AT1-). When the butyl side chain was removed and the amide functionality of 6,7-dimethoxyquinazoline-4-one was reduced to the free amine as in compounds of type (D, E, F and G) some better results were obtained, particularly compounds (42 and 110) were found to show the highest balanced activity (pA2 for α1 = 9.45 and 8.77 and AT1 = 8.36 and 8.60 respectively) on both the receptors.
Table 1 Actual and predicted activities (pA2 values for α1- and AT1-receptors) for the synthesized compounds (10–16, 24–29, 33–35, 38, 42–44, 50–53, 61–67 and 96–123)e
| Compound |
α1-Antagonistic activity (pA2) |
AT1-Antagonistic activity (pA2) |
| Actual |
Predicted |
Residuals |
Actual |
Predicted |
Residuals |
| Did not show any activity. Test set compounds for α1-antagonistic activity. Test set compounds for AT1-antagonistic activity. Test set compounds for both α1- and AT1-antagonistic activity. OL = compounds found as outliers in the pharmacophore modeling, NT = not taken for computational study due to absence of biological activity in the compound. |
| 10b |
6.97 |
6.54 |
0.43 |
7.00 |
6.98 |
0.02 |
| 11 |
6.42 |
6.08 |
0.34 |
5.53 |
5.40 |
0.13 |
| 12 |
6.40 |
6.33 |
0.07 |
6.92 |
6.80 |
0.12 |
| 13 |
7.13 |
6.69 |
0.44 |
6.44 |
6.30 |
0.14 |
| 15 |
5.50 |
5.44 |
0.06 |
6.01 |
6.00 |
0.01 |
| 16c |
5.90 |
5.32 |
0.58 |
5.76 |
5.42 |
0.34 |
| 24 |
6.06 |
6.08 |
−0.02 |
5.13 |
4.87 |
0.26 |
| 25b |
5.69 |
5.89 |
−0.20 |
5.62 |
5.29 |
0.33 |
| 26d |
5.27 |
5.73 |
−0.46 |
5.75 |
5.82 |
−0.07 |
| 27 |
4.53 |
4.74 |
−0.21 |
5.19 |
4.93 |
0.26 |
| 28 |
5.63 |
5.74 |
−0.11 |
4.90 |
5.01 |
−0.11 |
| 29 |
5.48 |
5.27 |
0.21 |
5.28 |
5.22 |
0.06 |
| 33 |
7.71 |
7.79 |
−0.08 |
7.41 |
7.25 |
0.16 |
| 34 |
6.33 |
6.49 |
−0.16 |
5.68 |
5.63 |
0.05 |
| 35 |
6.49 |
6.59 |
−0.10 |
5.91 |
5.71 |
0.20 |
| 38c |
6.82 |
7.07 |
−0.25 |
7.01 |
7.32 |
−0.31 |
| 42 |
9.45 |
8.77 |
0.68 |
8.36 |
8.70 |
−0.34 |
| 43 |
7.26 |
7.64 |
−0.38 |
6.54 |
5.84 |
0.70 |
| 44c |
7.09 |
7.45 |
−0.36 |
7.59 |
7.34 |
0.25 |
| 50 |
5.26 |
5.47 |
−0.21 |
6.15 |
6.08 |
0.07 |
| 51 |
3.01 |
3.68 |
−0.67 |
3.70 |
3.67 |
0.03 |
| 52 |
4.19 |
4.57 |
−0.38 |
3.22 |
3.22 |
0.00 |
| 53b |
5.93 |
6.78 |
−0.85 |
5.47 |
5.32 |
0.15 |
| 61b |
6.37 |
6.55 |
−0.18 |
6.27 |
7.31 |
−1.04 |
| 62 |
5.52 |
6.45 |
−0.93 |
4.87 |
5.23 |
−0.36 |
| 63 |
6.16 |
6.43 |
−0.27 |
5.38 |
4.74 |
0.64 |
| 64 |
6.33 |
6.98 |
−0.65 |
6.75 |
7.46 |
−0.71 |
| 65b |
7.02 |
6.57 |
0.45 |
6.27 |
6.57 |
−0.30 |
| 66 |
4.89 |
OL |
OL |
4.65 |
4.86 |
−0.21 |
| 67c |
6.78 |
6.37 |
0.41 |
7.23 |
6.97 |
0.26 |
| 96 |
6.49 |
5.98 |
0.51 |
6.78 |
6.44 |
0.34 |
| 97 |
6.76 |
6.75 |
0.01 |
6.09 |
6.25 |
−0.16 |
| 98 |
5.95 |
5.71 |
0.24 |
4.75 |
4.97 |
−0.22 |
| 99 |
5.66 |
5.85 |
−0.19 |
7.75 |
7.24 |
0.51 |
| 100c |
6.46 |
6.28 |
0.18 |
5.37 |
5.81 |
−0.44 |
| 101 |
a |
NT |
— |
a |
NT |
— |
| 102 |
2.14 |
2.32 |
−0.18 |
4.36 |
OL |
— |
| 103 |
a |
NT |
— |
6.61 |
6.47 |
0.14 |
| 104 |
a |
NT |
— |
6.34 |
6.58 |
−0.24 |
| 105 |
a |
NT |
— |
a |
NT |
— |
| 106 |
3.28 |
2.74 |
0.54 |
2.15 |
OL |
— |
| 107 |
6.93 |
OL |
— |
6.87 |
7.05 |
−0.18 |
| 108 |
4.55 |
4.32 |
0.23 |
5.22 |
OL |
— |
| 109 |
a |
NT |
— |
6.10 |
5.73 |
0.37 |
| 110 |
8.77 |
7.88 |
0.89 |
8.60 |
7.85 |
0.75 |
| 111 |
5.08 |
4.62 |
0.46 |
a |
NT |
— |
| 112 |
5.89 |
5.74 |
0.15 |
a |
NT |
— |
| 113 |
5.91 |
6.11 |
−0.20 |
6.49 |
6.40 |
0.09 |
| 114 |
5.91 |
6.74 |
−0.83 |
6.12 |
6.30 |
−0.18 |
| 115 |
7.19 |
6.99 |
0.20 |
a |
NT |
— |
| 116d |
7.50 |
6.58 |
0.92 |
6.58 |
6.31 |
0.27 |
| 117 |
4.64 |
4.45 |
0.19 |
a |
NT |
— |
| 118b |
6.32 |
6.83 |
−0.51 |
6.95 |
7.49 |
−0.54 |
| 119 |
7.44 |
7.74 |
−0.30 |
7.26 |
7.38 |
−0.12 |
| 120 |
a |
NT |
— |
6.23 |
6.24 |
−0.01 |
| 121 |
1.98 |
2.34 |
−0.36 |
3.03 |
OL |
— |
| 122b |
4.05 |
4.32 |
−0.27 |
5.41 |
5.12 |
0.29 |
| 123 |
a |
NT |
— |
a |
NT |
— |
Table 2 Actual and predicted activities (pA2 values for α1- and AT1-receptors) for the compounds reported19 earlier (124–138 and 3)

|
| Compound |
X/Ar |
α1-Antagonistic activity |
AT1-Antagonistic activity |
| Actual |
Predicted |
Residuals |
Actual |
Predicted |
Residuals |
| Test set compounds for α1-antagonistic activity. Test set compounds for AT1-antagonistic activity. |
| 124 |
–CN |
6.88 |
6.8 |
0.08 |
6.65 |
6.73 |
−0.08 |
| 125 |
–Tez (CHN4) |
7.10 |
7.27 |
−0.17 |
7.03 |
7.26 |
−0.23 |
| 126b |
–COOH |
7.45 |
7.16 |
0.29 |
6.14 |
5.50 |
0.64 |
| 127 |
–CN |
7.71 |
8.19 |
−0.48 |
7.41 |
7.62 |
−0.21 |
 |
| 128 |
–C6H5 |
9.89 |
10.2 |
−0.31 |
8.37 |
7.78 |
0.59 |
| 129 |
3-C6H4NO2 |
8.09 |
7.54 |
0.55 |
9.04 |
8.78 |
0.26 |
| 130a |
4-C6H4COOMe |
6.86 |
7.59 |
−0.73 |
10.64 |
9.86 |
0.78 |
| 131 |
4-C6H4NO2 |
9.38 |
9.22 |
0.16 |
7.64 |
7.17 |
0.47 |
| 132 |
–C10H7 |
8.37 |
8.41 |
−0.04 |
7.07 |
7.29 |
−0.22 |
| 133 |
3-COOH |
7.09 |
7.15 |
−0.06 |
7.04 |
7.90 |
−0.86 |
| 134a |
4-Me |
8.53 |
7.82 |
0.71 |
7.65 |
7.79 |
−0.14 |
| 135 |
–H |
8.74 |
8.35 |
0.39 |
3.31 |
4.33 |
−1.02 |
| 136 |
2-F |
10.51 |
10.99 |
−0.48 |
5.52 |
5.28 |
0.24 |
| 137a |
–Me |
9.23 |
9.03 |
0.20 |
5.75 |
5.91 |
−0.16 |
| 138 |
–Et (C2H5) |
10.41 |
9.56 |
0.85 |
5.08 |
5.88 |
−0.80 |
| 3 |
2-CN |
10.10 |
10.46 |
−0.36 |
8.83 |
8.91 |
−0.08 |
Type A compounds (10–15) having electron withdrawing groups on benzyl ring showed moderate but somewhat better activity than compound (16) having electron releasing group. When the position of the butyl chain was changed from 2nd to 3rd and the benzyl ring replaced with anilinomethyl moiety, the effect of different (i.e. electron withdrawing, electron releasing and heterocycles) substituents in compounds of type B (24–29) was assessed, it was found out that the compounds were equally active as compounds of type A. Replacement of methylanilino moiety with substituted piperazines as in compounds of type C (33–35) offered still more improvement in antagonistic potencies on both (α1 and AT1) the proposed receptors; compound (33) showed balanced moderate to high (pA2 for α1 = 7.71 and AT1 = 7.41 respectively) antagonistic activity. Replacement of 4-keto by the 4-amino group and attaching anilino/methylanilino moiety in place of the piperazine ring as in compounds of type D (38, 61–67) yielded almost equally potent compounds to compounds of type A.
Restoring piperazine ring at 2nd position of 4-amino-6,7-dimethoxyquinazoline moiety from anilino/methylanilino, with or without a methylene spacer offered potent compounds of type E (42–44 and 96–97) with balanced activity. Cyano group at 2nd position of phenyl ring of 2-(piperazin-1-yl)-6,7-dimethoxy-4-aminoquinazoline moiety yielded the most active (pA2 for α1 = 9.45 and AT1 = 8.36) compound (42) among them.
Compounds (98–123) having a methylene spacer between the 4-amino-6,7-dimethoxyquinazoline moiety and the piperazine ring and a keto spacer (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) between the piperazine ring and the phenyl ring (98–107) yielded compounds with variable potencies. Compound (107) showed balanced activity (pA2 for α1 = 6.93 and AT1 = 6.87) on both the receptors. Compounds (98–100) were also found to be moderately (pA2 = 4–7) active on both the receptors but the activities were not balanced. Compounds (103–104) having dichloro and dimethoxy groups at 2nd and 3rd positions of phenyl ring respectively, were observed to be moderately active at AT1-receptor but inactive at α1-receptor. Compounds (101 and 105) were found to be inactive on both the receptors.
O) between the piperazine ring and the phenyl ring (98–107) yielded compounds with variable potencies. Compound (107) showed balanced activity (pA2 for α1 = 6.93 and AT1 = 6.87) on both the receptors. Compounds (98–100) were also found to be moderately (pA2 = 4–7) active on both the receptors but the activities were not balanced. Compounds (103–104) having dichloro and dimethoxy groups at 2nd and 3rd positions of phenyl ring respectively, were observed to be moderately active at AT1-receptor but inactive at α1-receptor. Compounds (101 and 105) were found to be inactive on both the receptors.
Compounds (121–123) in which phenyl(piperazinyl)methanone moiety was replaced with 2-phenyl(piperazinyl)ethanone, failed to produce satisfactory results on both the receptors. Although compound (122) showed balanced activity but it was no where comparable to the standard drugs (terazosin and losartan). Compound (123) was found to be inactive on both the receptors.
On the other hand replacement of phenyl(piperazinyl)methanone moiety with substituted benzylpiperazine groups, the resulting derivatives (108–116) offered better results in the preliminary screening. Compound (110) (pA2 for α1 = 8.77 and AT1 = 8.60) showed almost equal antagonistic potency to the standard drugs terazosin and losartan.
Compounds (108, 113–114 and 116) also showed dual antagonism but the potencies were ranging from poor to moderate. Compound (109) having strong electron withdrawing group (CF3) at 2nd position of phenyl ring was found to be moderately active at AT1-receptor but inactive at α1-receptor. Compounds (111, 112, 115 and 117) were found to be moderately active at α1-receptor but were inactive at AT1-receptor. Compound (119) having benzhydryl moiety showed balanced antagonistic activity at both the receptors but showed lower potencies than terazosin for α1- and losartan for AT1-receptor. Compound (118) having 1-naphthyl moiety showed similar type of results as shown by compound (119).
Putting an alkyl substituent on the piperazine ring in place of aryl ones or replacing the piperazinyl aryl groups with some other heterocyclic rings as was the case with compounds of type G (50–53), reduced potency on both the receptors was obtained, although compound (50) showed somewhat moderate potency amongst them.
Thus, two compounds amongst all of the test compounds of the series were found to be the most potent dual antagonists in the in vitro tissue experiments. As shown in Fig. 1, both of these compounds (42 and 110) showed competitive antagonism against phenylephrine and angiotensin II. A rightward parallel shift was observed in the dose response curve after addition of compounds (42 and 110). The results demonstrated competitive dual antagonism by compounds (42 and 110) on α1- as well as AT1-receptors.
 |
| | Fig. 1 Dose response plots of compounds 42 (A) and 110 (B) against phenylephrine on rat aortic strips (n = 4). Dose response plots of compounds 42 (C) and 110 (D) against AII on rat aortic strips (n = 4). A rightward parallel shift can be observed in presence of compounds (42 and 110) against phenylephrine and AII mediated responses. | |
2.3. Invasive blood pressure response
Preliminary screening results showed that the two compounds (42 and 110) have promising antagonistic activity on both of the stated receptors (pA2 for α1 = 9.45 and 8.77 and AT1 = 8.36 and 8.60 respectively). So, it was planned to revalidate the in vitro results into in vivo animal model for both of the compounds (42 and 110). Since invasive blood pressure (IBP) measurement provides direct assessment of antihypertensive activity of the investigational compounds, it has been utilized as a gold standard technique for evaluation of potential antihypertensive compounds.28–31 Both the compounds (42 and 110) were evaluated against phenylephrine (6 μg kg−1) and AII (6 μg kg−1) induced rise in BP in the rat model. In our earlier finding, we have reported that fall in blood pressure mediated by prazosin was not only because of α1 antagonism (pA2 for α1 = 8.91), but also due to simultaneous blockade of AT1-receptor32 (pA2 for AT1 = 8.26). Thus, for the present study terazosin was chosen as a standard α1 antagonist instead of prazosin and losartan as a standard AT1-receptor antagonist against phenylephrine and AII mediated pressor responses respectively.
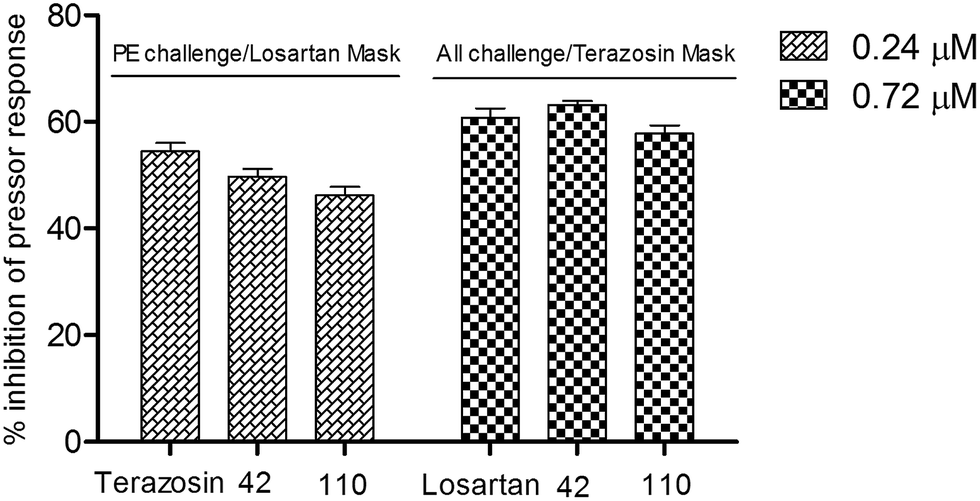
For the purpose of comparison, the dose levels of the agonists and antagonists were maintained as per our previous study.19 Despite having almost equal or higher potency then terazosin in the in vitro experiments, neither of the selected compounds (42 and 110) could replicate the response of the standard α1-blocker terazosin in the in vivo animal model (Fig. 2). This is probably due to the distribution of these test compounds (42 and 110) on both the receptors, as these compounds are dual antagonists while the standard terazosin inhibits only α1-receptor. So the experiments were repeated by masking one receptor at a time and evaluating the test compounds on the other receptor. Terazosin (0.72 μM) was used for masking of α1-receptor and the test compounds (42 and 110) were evaluated for their antagonism of AII response while losartan (3.6 μM) was used for masking of AT1-receptor and the test compounds evaluated for their antagonism of phenylephrine response on α1-receptor, as described in our previous report.19
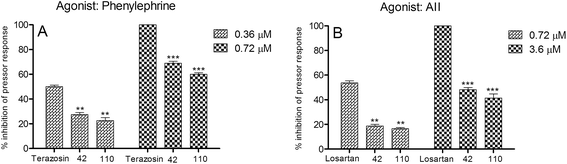 |
| | Fig. 2 (A) Inhibition of mean arterial pressor response of phenylephrine in animals (n = 4) previously dosed with compounds (42 and 110), or terazosin at equimolar levels. (B) Inhibition of mean arterial pressor response of AII in animals (n = 4) previously dosed with compounds (42 and 110), or losartan at equimolar levels. Results are expressed as mean ± SEM values. **p < 0.001, ***p < 0.0001. | |
Under masking conditions, both of the test compounds (42 and 110) were found to be almost equipotent to the two standard drugs, terazosin as α1-antagonist and losartan as AT1-antagonist (Fig. 3) supporting the observations of the in vitro experiments of dual antagonist nature of the synthesized compounds.
 |
| | Fig. 3 In vivo pressure response inhibition of phenylephrine and AII by compounds (42 and 110) in normotensive rats (n = 4) using losartan (3.6 μM) and terazosin (0.72 μM) respectively as masking agents. Results are expressed as mean ± SEM values. PE = phenylephrine. | |
2.4. Molecular modeling studies
It was planned to perform molecular modeling studies for identifying the structural features for the dual activity of the compounds under study. For this purpose synthesized compounds reported here and in our earlier publication19 constituted a sizable database which were used for deriving significantly predictive ligand based pharmacophore and atom based 3D-QSAR models. The receptor independent pharmacophore model (i.e. ligand based pharmacophore model) was derived that was supported by 3D-QSAR model. The predictive ability of the developed model was validated by internal test set as well as by using two marketed standard drugs as external test compounds. It was also planned to perform docking studies on some selected compounds using self developed homology models of α1- and AT1-receptors. This docking study was planned mainly to understand the ligand receptor interactions and to validate the developed pharmacophoric model by comparing it with the ligand–receptor docking interaction features. The results so obtained strongly supported the validity of the developed homology model as well as the pharmacophore and 3D-QSAR models. All the results obtained in the study are described in the following sections. For development of the pharmacophore model, compounds with activity above 8 were considered as active and below 5.5 were considered as inactive.
2.4.1. Pharmacophore and 3D-QSAR modeling studies for α1-receptor antagonists. The compounds used for this study are given in Table 1 along with their biological activity. PHASE model of Schrodinger was used for these studies. PHASE works on tree-based partitioning algorithm systematically identifying the spatial arrangement of functional groups essential for the biological activity from a set of high affinity ligands. Typically, PHASE offers a pharmacophore model using a combination of six structural features, namely hydrogen bond acceptor (A), hydrogen bond donor (D), hydrophobic (H), negatively ionizable (N), positively ionizable (P) groups and aromatic rings (R). Using the dataset for α1-receptor antagonists, various models were obtained. The models so obtained were evaluated by the scoring alignment of ‘actives’ against a reference ligand by using default settings for ‘score actives’ to recognize the pharmacophore features from each box that resulted into the best alignment of the active ligands. The models were ranked according to the survival values for ‘active’ and ‘inactive’ compounds and evaluated on the basis of ‘survival’ and ‘survival-inactive’. The best pharmacophore model for α1-receptor antagonists was obtained with five pharmacophoric features (ADHHR). The inter-site distances and angles of pharmacophoric groups are depicted in Fig. 4 with the best fit and second most potent compound (138) aligned on the model. These features represent the optimum requirement for a compound to show α1-receptor antagonistic activity.
 |
| | Fig. 4 Best pharmacophore model (ADHHR) for α1-receptor antagonists aligned on best fit compound (138) with (A) intersite distance in Å; (B) angles between pharmacophore features. | |
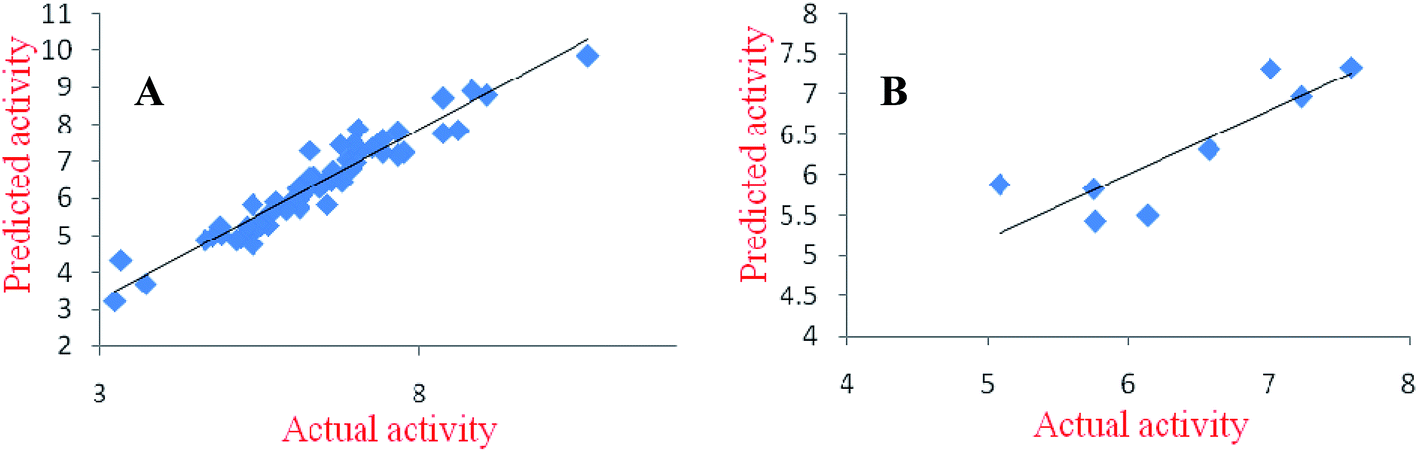
In the best pharmacophore model (ADHHR), two compounds (66 and 107) were found to be outliers from the data set of 67 compounds. Excluding these two compounds a 3D-QSAR model was developed. The dataset of 65 compounds was divided into a training set of 53 and test set of 12 molecules. The model was statistically developed by using partial least square (PLS) method. The atom based 3D-QSAR model was utilized for predicting the biological activity of the training as well as the test set of compounds. Statistical parameters (like R2, Q2, SD, RMSE, F-value and Pearson-R) obtained for the model validated its reliability. Here, R2 explains the regression coefficient of prediction for training set whereas same for the test set is explained by Q2. Pearson-R represents the correlation between the predicted and the actual activities for the test set. The scattered plots for actual and predicted activity for training and test set of compounds are shown in Fig. 5.
 |
| | Fig. 5 The scattered plots for actual and predicted activity for training set (A); and test set (B) of α1 model. | |
The 3D-QSAR volume occlusion maps represent the features essential for the ligand–receptor interactions. The occlusion maps are represented for PLS factor 5 by blue and red colors representing favorable and unfavorable interactions respectively. The occlusion maps (donor, hydrophobic and electronegative) with the best fit compound (138) for the developed 3D-QSAR model are shown in Fig. 6. In Fig. 6A, the donor map explains the high activity of compounds like (110, 136 and 138) as the amine group aligns to the hydrogen bond donor region. The low activity of compounds (15, 27 and 29) is also explained by the occlusion maps. The hydrophobic/non-polar map for the best fit compound is shown in Fig. 6B. The map explains the high potency of compounds (110, 136 and 138). The methoxy groups, hydrophobic part of piperazine ring as well as the aromatic and alkyl substituents on piperazine ring occupy the positions in blue regions. These observations were also supported by the docking study of compound (110), wherein the methoxyl group on the aromatic ring appears in the hydrophobic cadge of Phe61, Phe65 and Trp71, whereas the aromatic ring attached to the piperazine group was observed to be aligned with the hydrophobic cadge of Leu174, Phe267 and Phe268. These contours also explain the poor activity of compounds (16, 50, 102 and 121) as the majority of the non-polar part of these ligands is oriented towards the red contours, and in docking, orientations of these were observed to be deviating from the hydrophobic sites (data not shown). The electron withdrawing contour map, as shown in Fig. 6C, explains the high activity of compounds (110, 136 and 138) as the amine and the nitrogen of piperazine rings are in blue region of the contour map. The high activity of compounds like 42 and 131 was also explained very well as the cyano and nitro groups on the aromatic rings of the respective compounds lied in red contour of the occlusion map.
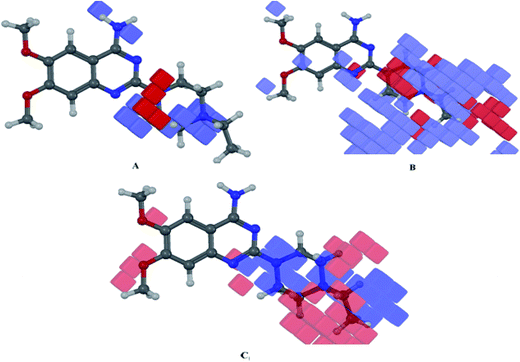 |
| | Fig. 6 3D-QSAR visualization for compound (138); H-bond donor (A), hydrophobic (B) and electron-withdrawing (C) components. | |
To strengthen the validity of the developed computational models, the models were further evaluated by using various statistical methods previously reported by different groups as described in the experimental section. The R2m (test) value of 0.593 (>0.5) suggested that the developed model was valid for any new dataset as well. To further prove its reliability, α1-receptor antagonistic activity of terazosin and prazosin, as compounds outside the domain of the data set, was predicted using the developed model. The predicted activity for terazosin [S-isomer: 8.26 and R-isomer: 8.45] using the developed model was found to be quite close to the experimental value (pA2 = 8.65). Similarly, the inhibitory activity for prazosin for α1-receptor was predicted to be 8.50, which was very close to the experimentally determined pA2 value of prazosin for α1-receptor (pA2 = 8.08). To determine the sensitivity, accuracy, specificity, positive prediction value and negative prediction value, the dataset was divided as per the division rule explained in the experimental section. As per this division, compounds with pA2 values < 6.5 were considered as inactives and compounds with pA2 values ≥ 6.5 as actives. The sensitivity value of the model indicates that 90% of the compounds were correctly predicted as ‘active’ while the specificity value suggested that 82.8% of the compounds were correctly predicted as ‘non-active’. The accuracy level for the developed model was 86.1%. All these statistical predictions along with comparison of the experimentally determined activity and the activity predicted using the developed model for the standard drugs, suggested that the developed models are robust and possessed good prediction ability which could be used for designing of novel dual acting α1- and AT1-antagonists.
2.4.2. Pharmacophore and 3D-QSAR modeling studies for AT1-receptor antagonists. On similar lines to the modeling studies for α1-receptor antagonists, modeling studies for AT1-receptor antagonists were also performed. A five point pharmacophore model (ADHRR) was developed for AT1-receptor antagonists. The inter-site distances and angles among the pharmacophoric features, aligned on the best fit and the second most active compound (3) are depicted in Fig. 7. It is worth noting that all the pharmacophoric features (ADHHR) of the developed α1-receptor antagonists are restricted to the quinazoline moiety (Fig. 4) while for AT1-receptor antagonists (ADHRR), one of the features is shifted to the aromatic ring (R) of the side chain (Fig. 7). In the best developed pharmacophore model (ADHRR) four compounds (102, 106, 108 and 121) of the data set were found to be outliers. Out of the remaining 63 compounds, 55 compounds were retained in training set and 8 compounds in the test set. Atom-based 3D-QSAR model was developed by applying PLS algorithm. The scattered plots for the actual and the predicted activities for the training and test set compounds are shown in Fig. 8.
 |
| | Fig. 7 Best pharmacophore model (ADHRR) for AT1-receptor antagonists aligned on best fit compound (3) with (A) intersite distance in Å; (B) angles between pharmacophore features. | |
 |
| | Fig. 8 The scattered plots for actual and predicted activity for training set (A); and test set (B) of AT1 model. | |
Contour maps on the best fit compound (3) are analyzed for this 3D-QSAR model and are represented in Fig. 9. The hydrogen bond donor occlusion map as shown in Fig. 9A, explains the high activity of compounds (129, 130 and 3) as the amino group was observed to be in the favorable region of the contour map. Docking study also supports these pharmacophoric features as hydrogen bonding is observed between –NH2 with Ser105. This map also explained lesser potency of compounds like 66 and 135 as the hydrogen bond donor groups were oriented away from the favorable region. The hydrophobic occlusion map (Fig. 9B) represented the high activity of compounds like 129, 130 and 3. Poor activity of some compounds was also explained as the aliphatic and aromatic hydrophobic groups attached to the piperazine ring in the compounds fall into the polar red region. This observation was also supported by the docking study. Fig. 9C represents the electron withdrawing occlusion map. The cyano group in compound (3) and ester group in compound (130) align into the favorable region and thus the compound exhibits high activity.
 |
| | Fig. 9 3D-QSAR visualization for 3; H-bond donor (A), hydrophobic (B) and electron-withdrawing (C) components. | |
On similar lines as described for the α1-receptor antagonists, the dataset of AT1-receptor antagonists was divided into two groups, the group with pA2 values ≥ 6.3 were considered as ‘actives’ and the other with pA2 values < 6.3 as ‘inactives’ to determine the additional validation parameters like sensitivity, accuracy, specificity, positive prediction value and negative prediction value. The sensitivity level of 82.7%, specificity of 90.9% and accuracy level of 87% proved the robustness of the developed model. The R2m (test) value for the model was found to be 0.655 which was above 0.5. The statistical results for both the developed models are shown in Table 3. The model was further validated by predicting the pA2 value for the two marketed drugs terazosin and prazosin. For terazosin the predicted AT1-receptor antagonist activity (predicted pA2 for S-isomer = 6.27; R-isomer = 6.93) was close to the experimentally determine one (pA2 = 6.39). For prazosin the experimentally determined pA2 activity was 8.26 and the predicted activity from the model was 7.66. These predictions and the results of the statistical values strongly support the predictive ability of the developed models.
Table 3 Statistical results for common pharmacophore and 3D-QSAR models developed for α1- and AT1-receptor antagonism generated by PLS
| Model for α1-receptor antagonists |
Model for AT1-receptor antagonists |
| Training set |
Test set |
Training set |
Test set |
| PLS factor = 5 |
PLS factor = 5 |
| n = 53 |
n(test) = 12 |
n = 55 |
n(test) = 8 |
| R2 = 0.953 |
Q2 = 0.829 |
R2 = 0.918 |
Q2 = 0.715 |
| SD = 0.427 |
RMSE = 0.552 |
SD = 0.415 |
RMSE = 0.428 |
| F = 193.2 |
Pearson-R = 0.915 |
F = 110.1 |
Pearson-R = 0.853 |
| P = 3.984 × 10−30 |
P = 1.97 × 10−31 |
| Validation parameters |
Validation parameters |
| R2m (test) |
0.593 |
R2m (test) |
0.655 |
| R2m (Loo) |
0.905 |
R2m (Loo) |
0.916 |
| R2m (overall) |
0.876 |
R2m (overall) |
0.907 |
| k |
1.006 |
k |
1.043 |
| k′ |
0.987 |
k′ |
0.956 |
| Sensitivity |
0.900 |
Sensitivity |
0.827 |
| Specificity |
0.828 |
Specificity |
0.909 |
| Accuracy |
0.861 |
Accuracy |
0.870 |
| PPV |
0.818 |
PPV |
0.888 |
| NPV |
0.906 |
NPV |
0.857 |
| MCC |
0.727 |
MCC |
0.741 |
| R2 − R02/R2 |
0.100 |
R2 − R02/R2 |
0.063 |
| R2 − R0′2/R2 |
0.009 |
R2 − R0′2/R2 |
0.008 |
2.5. Docking studies
Docking studies were performed to understand the ligand–receptor interactions by using homology models developed for the α1- and AT1-receptors. The docking interactions of the active ligands under study on the two receptors are discussed here.
2.5.1. α1-Receptor. As the amino acid sequence was truncated by initial 21 amino acids, the interacting residue numbering is mentioned accordingly. Interactions with the active ligands (3 and 110) were analyzed by the docking study. In case of ligand (3), the protonated NH of piperazine forms a salt bridge with Asp85. One of the methoxy groups of dimethoxyquinazoline ring formed H-bonding with Ser62. The aromatic ring of quinazoline offered further stability to the receptor–ligand complex by showing π–π stacking with Trp81 (Fig. 10A). In case of ligand (110), a similar type of salt bridge was observed with Asp85. Additionally, the o-methylbenzyl group was observed to be present into the hydrophobic pocket of Cys89, Phe267, Phe268, Met271 and Pro272. The quinazoline part was found to be stabilized by hydrophobic interactions with Phe65, Trp71 and Phe291 (Fig. 10B).
 |
| | Fig. 10 Docking interactions of 3 (A) and 110 (B) with the α1-receptor. | |
2.5.2. AT1-Receptor. The docking interactions of compounds (110 and 130) were studied similarly within the active site of AT1-receptor. The aromatic ring of quinazoline in 130 was found to be stabilized by hydrophobic interactions with Leu112, Tyr113 and Trp253. The anilino part of the ligand lent stability to the receptor–ligand complex by forming π–π stacking interaction with Trp84. The amino group of quinazoline ring offered further stability by having H-bonding with Ser105 and Ser109 (Fig. 11A). In the case of 110, the π-cation interaction of the aromatic ring of quinazoline and H-bonding of the amino group with His256 offered stability to the complex. But the orientation of o-methylbenzyl group towards Arg167 may be causing non-favorable interactions at this end as one part is polar and the other is hydrophobic in nature (Fig. 11B).
 |
| | Fig. 11 Docking interaction of 130 (A) and 110 (B) with the AT1-receptor. | |
After docking the antagonists [compounds (3 and 110) for α1-receptor and compounds (110 and 130) for AT1-receptor] in the active sites of the respective homology models of the receptors, the docked-conformations of the molecules were retrieved from the active sites and matched with the conformations obtained from the pharmacophore modeling, in order to further assess the receptor-independent and receptor-dependent conformational variations. The superposed conformations were matching with each other within acceptable limits (RMSD 1.89 ± 0.21 Å). These observations further supported the validity of both of the pharmacophore models for dual acting α1- and AT1-antagonists.
3. Conclusion
A series of 6,7-dimethoxyquinazoline derivatives (10–16, 24–29, 33–35, 38, 42–44, 50–53, 61–67 and 96–123) have been synthesized and evaluated as dual acting α1- and AT1-receptor antagonists. In the present study we focused on the strategy of obtaining balanced activity in the compounds i.e. equal level of antagonism at α1- as well as AT1-receptors. Compound (42) having 2-cyanophenylpiperazine substituent at the 2nd position of 4-amino-6,7-dimethoxyquinazoline, showed the highest potency and balanced modulation of both the receptors among the synthesized compounds. Compound (110) was found to be almost equally active as (42) on AT1-receptor but it showed slightly less potency on α1-receptor. Balanced activity obtained for compound (33) may be due to the presence of cyano group at 2nd position. Compounds (44) and (119) having a phenyl group substituted on the carbon of benzylpiperazine ring also showed balanced activity. Compounds (98–107) and (121–123) having 1-phenyl-1-(piperazinyl)methanone and 2-phenyl-1-(piperazinyl)ethanone moieties respectively were found to have poor activity. It was shown earlier that compounds bearing electron withdrawing groups on the aromatic ring of the side chain at 2nd position of 4-amino-6,7-dimethoxyquinazoline, as in compound (3), showed potent dual antagonistic activity. The current studies revealed that a mild electron releasing group on the aromatic moiety at 2nd position of 4-amino-6,7-dimethoxyquinazoline scaffold can also render sufficient potency to the resulting compounds as shown by compound (110). Two potential resultants (42 and 110) from the in vitro studies yielded almost equal dual potency to the standard α1-receptor antagonist prazosin and AT1-receptor antagonist losartan when studied in the in vivo animal model. Using the available data of biological activity in hand for the synthesized compounds, highly predictive pharmacophore and 3D-QSAR models were developed. Further, these developed models were validated by different well described and accepted validation parameters. In order to further validate the models the activity of two marketed standard drugs were also predicted by using these two developed models and the results so obtained fully explained the predictive accuracy of the developed models. To further extend the validation of the developed ligand based pharmacophore and QSAR models, homology models for α1- and AT1-receptors were developed and used for docking studies. Conformations of the antagonists obtained after docking of the molecules in the active sites of both of the homology models i.e. α1-receptor and AT1-receptor, matched with the pharmacophoric features obtained from pharmacophore modeling. Thus, the docking studies further validated the developed models for the dual antagonists.
4. Experimental section
4.1. General methods
Melting points of all the synthesized compounds were determined in open glass capillaries using a Veego make silicon oil bath-type melting point apparatus and are uncorrected. The IR spectra were recorded using KBr disc method on a Bruker FT-IR, Model ALPHA-T. 1H-NMR and 13C-NMR spectra were recorded in CDCl3 or DMSO-d6 solutions on a Bruker Avance II 400 NMR spectrometer with TMS (tetramethylsilane) as the internal standard. Chemical shifts are reported in parts per million (ppm) relative to TMS and multiplicities are given as s (singlet), d (doublet), t (triplet) and m (multiplet). Mass spectra were recorded using a Thermo Fisher mass spectrometer with EI as ion source or an Advion mass spectrometer with ESI as ion source. Compounds were purified through chromatographic separations using silica gel (100–200 mesh) as well as neutral alumina as stationary phases. Purity of the compounds and completion of reactions were monitored by thin layer chromatography (TLC) on silica gel G plates, visualizing with ultraviolet light or iodine vapors. Purity was determined by HPLC analysis and was found above 98% for all the compounds. Elemental analysis was performed on Thermo Scientific flash-2000 elemental analyzer.
The intermediates (4, 17, 36, 45),19 (71–77, 81, 83, 84, 92, 94),20,21,23 (70, 78, 79)20,21 (93, 95)20,22 (80, 82, 86, 87)20,23,25 (85, 88, 90, 91)20,24,26 (92)20,27 were synthesized as per earlier reported procedures while compounds (5–9, 18–23, 30–32, 37, 39–41, 46–49, 54–60, 68, 69) were obtained from commercial sources.
4.1.1. Methyl-3-[(2-butyl-6,7-dimethoxy-4-oxoquinazolin-3(3H)-yl)methyl]benzoate (10). Method A: 2-butyl-6,7-dimethoxyquinazolin-4(3H)-one (4) (0.5 g, 1.9 mM), methyl-3-bromomethylbenzoate (5) (0.87 g, 3.8 mM) and flame dried potassium carbonate (0.52 g, 3.8 mM) were taken in dry acetone. The reaction mixture was stirred at RT and monitored by TLC. After completion of reaction, the reaction mixture was poured into ice-cold water, filtered off the precipitate and washed the precipitate with cold water. The dried precipitate was recrystallized from dichloromethane–methanol to afford the desired compound (10) (0.45 g, 58%) as white crystals, mp = 178–80 °C. IR (KBr): 3129, 3006, 2958, 2933, 2871, 1669, 1612, 1568, 1500, 1466, 1404, 1273, 1245, 1032, 785 cm−1. Anal. calcd for C23H26N2O5: C, 67.30; H, 6.38; N, 6.82. Found: C, 67.52; H, 6.27; N, 6.73%.
4.1.2. Methyl-4-[(2-butyl-6,7-dimethoxy-4-oxoquinazolin-3(3H)-yl)methyl]benzoate (11). Following method A methyl 4-bromomethylbenzoate (6) (0.87 g, 3.8 mM) afforded compound (11) (0.43 g, 55%), mp = 187–89 °C. IR (KBr): 3127, 2998, 2970, 2944, 2838, 1720, 1654, 1612, 1504, 1435, 1284, 1233, 1113, 1013, 844, 750 cm−1. 1H-NMR (DMSO-d6): δ 7.95–7.93 (d, 2H, J = 8.4 Hz), 7.46 (s, 1H), 7.30–7.28 (d, 2H, J = 8.4 Hz), 7.11 (s, 1H), 5.46 (s, 2H), 3.92 (s, 3H), 3.88 (s, 3H), 3.83 (s, 3H), 2.69–2.65 (t, 2H, J = 7.5 Hz), 1.67–1.60 (m, 2H), 1.33–1.24 (m, 2H), 0.83–0.79 (t, 3H, J = 7.5 Hz). Anal. calcd for C23H26N2O5: C, 67.30; H, 6.38; N, 6.82. Found: C, 67.45; H, 6.29; N, 6.75%.
4.1.3. 4-[(2-Butyl-6,7-dimethoxy-4-oxoquinazolin-3(3H)-yl)methyl]benzonitrile (12). Following method A 4-bromomethylbenzonitrile (7) (0.75 g, 3.8 mM) afforded compound (12) (0.42 g, 58%), mp = 214–16 °C. IR (KBr): 3068, 3006, 2958, 2838, 2229, 1658, 1611, 1503, 1402, 1266, 1245, 1013, 847, 784 cm−1. Anal. calcd for C22H23N3O3: C, 70.01; H, 6.14; N, 11.13. Found: C, 70.23; H, 6.24; N, 10.95%.
4.1.4. 2-Butyl-6,7-dimethoxy-3-(3-nitrobenzyl)quinazolin-4(3H)-one (13). Following method A 3-nitrobenzyl bromide (8) (0.80 g, 3.8 mM) afforded compound (13) (0.46 g, 61%), mp = 160–62 °C. IR (KBr): 3088, 2961, 2941, 2874, 1665, 1613, 1590, 1530, 1503, 1444, 1352, 1272, 1208, 1000, 781 cm−1. Anal. calcd for C21H23N3O5: C, 63.46; H, 5.83; N, 10.57. Found: C, 63.19; H, 5.68; N, 10.63%.
4.1.5. 2-Butyl-6,7-dimethoxy-3-(4-nitrobenzyl)quinazolin-4(3H)-one (14). Following method A 4-nitrobenzyl bromide (9) (0.80 g, 3.8 mM) offered compound (14) (0.47 g, 63%), mp = 178–80 °C. IR (KBr): 2957, 2941, 2871, 1657, 1610, 1501, 1402, 1345, 1263, 1002, 733 cm−1. 1H-NMR (DMSO-d6): δ 8.21–8.19 (d, 2H, 8.7 Hz), 7.50 (s, 1H), 7.44–7.42 (d, 2H, 8.7 Hz), 7.11 (s, 1H), 5.52 (s, 2H), 3.97 (s, 3H), 3.93 (s, 3H), 2.72–2.68 (t, 2H, J = 7.5 Hz), 1.73–1.68 (m, 2H), 1.40–1.34 (m, 2H), 0.90–0.86 (t, 3H, J = 7.5 Hz). Anal. calcd for C21H23N3O5: C, 63.46; H, 5.83; N, 10.57. Found: C, 63.39; H, 5.75; N, 10.73%.
4.1.6. 4-[(2-Butyl-6,7-dimethoxy-4-oxoquinazolin-3(3H)-yl)methyl]benzoic acid (15). Aqueous sodium hydroxide solution (30%, 2.5 mL) was added in the solution of compound (11) (0.5 g, 1.2 mM) in methanol (25 mL) and refluxed for 30 minutes. After recovering excess amount of methanol the reaction mixture was poured into ice-cold water and acidified with dil. HCl (pH 5.5–6.0). Precipitate so obtained was filtered, washed several times with cold water and dried. The dried precipitate was recrystallized from DCM–methanol to afford the desired compound (15) (0.35 g, 74%) as white crystals, mp = 240–42 °C (dec.). IR (KBr): 3414, 3234, 2970, 1671, 1639, 1616, 1502, 1403, 1255, 1013, 756 cm−1. 1H-NMR (DMSO-d6): δ 8.23–8.21 (d, 2H, J = 8.7 Hz), 7.46 (s, 1H), 7.43–7.41 (d, 2H, J = 8.7 Hz), 7.12 (s, 1H), 5.51 (s, 2H), 3.93 (s, 3H), 3.87 (s, 3H), 2.70–2.66 (t, 2H, J = 7.0 Hz), 1.69–1.62 (m, 2H), 1.35–1.23 (m, 2H), 0.86–0.83 (t, 3H). Anal. calcd for C22H24N2O5: C, 66.65; H, 6.10; N, 7.07. Found: C, 66.42; H, 6.23; N, 7.23%.
4.1.7. 3-(4-Aminobenzyl)-2-butyl-6,7-dimethoxyquinazolin-4(3H)-one (16). A solution of 2-butyl-6,7-dimethoxy-3-(4-nitrobenzyl)quinazolin-4(3H)-one (14) (0.5 g, 1.3 mM) in methanol (20 mL) was refluxed on a water bath. Iron powder (0.6 g, 10.4 mM) and a solution of sodium chloride (0.55 g, 10.4 mM) in water (15 mL) were added portion-wise (in 4 parts at an interval of 45 minutes) to the refluxing solution. Refluxing was continued for 7–8 hours and the solution was filtered through filtering aid and washed with hot methanol (2 × 10 mL). The filtrate was concentrated under reduced pressure to remove excess of methanol and the resulting aqueous solution was diluted with water (25 mL), basified with sodium bicarbonate (10% aq. solution) and extracted with chloroform (3 × 50 mL). The combined chloroform layer was dried and concentrated to get brown colored residue which was dried under vacuum to afford compound (16) (0.38, 79%), mp > 280 °C. Rf 0.23 (hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate, 12:8). IR (KBr): 3414, 3159, 3007, 2959, 2872, 1659, 1614, 1497, 1436, 1400, 1249, 1138, 1001, 785 cm−1. Anal. calcd for C21H25N3O3: C, 68.64; H, 6.86; N, 11.44. Found: C, 68.39; H, 6.59; N, 11.32%.
:ethyl acetate, 12:8). IR (KBr): 3414, 3159, 3007, 2959, 2872, 1659, 1614, 1497, 1436, 1400, 1249, 1138, 1001, 785 cm−1. Anal. calcd for C21H25N3O3: C, 68.64; H, 6.86; N, 11.44. Found: C, 68.39; H, 6.59; N, 11.32%.
4.1.8. N-{3-[(3-Butyl-3,4-dihydro-6,7-dimethoxy-4-oxoquinazolin-2yl)methylamino]phenyl}methanesulfonamide (24). Method B: 3-butyl-2-chloromethyl-6,7-dimethoxyquinazolin-4(3H)-one (17) (0.5 g, 1.7 mM), 3-methanesulfonamidoaniline (18) (0.49 g, 2.6 mM), flame dried cesium carbonate (0.55 g, 1.7 mM) and dry DMF (2 mL) were taken in Rb flak (50 mL). The reaction mixture was stirred overnight and poured into ice-cold water (20 mL). The precipitate so obtained was filtered and washed several times with water. The solid residue was recrystallized from methanol to afford compound (24) (0.47 g, 60%), mp = 160–62 °C (dec.). IR (KBr): 3445, 3361, 2957, 2871, 1678, 1607, 1501, 1332, 1242, 1208, 1148, 1082, 786 cm−1. 1H-NMR (CDCl3): δ 7.58 (s, 1H), 7.13–7.09 (m, 1H), 6.93 (s, 1H), 6.90 (s, 1H), 6.89–6.88 (m, 1H), 6.61–6.58 (m, 1H), 5.00 (s, 2H), 4.15–4.11 (t, 2H, J = 7.6 Hz), 3.99 (s, 3H), 3.98 (s, 3H), 3.73 (bs, 2H), 3.23 (s, 3H), 1.71–1.67 (m, 2H), 1.48–1.42 (m, 2H), 0.99–0.96 (t, 3H, J = 7.6 Hz). MS (EI) m/z: 460.1 [M]+. Anal. calcd for C22H28N4O5S: C, 57.37; H, 6.13; N, 12.17. Found: C, 57.19; H, 6.38; N, 12.02%.
4.1.9. N-{4-[(3-Butyl-3,4-dihydro-6,7-dimethoxy-4-oxoquinazoline-2yl)methylamino]phenyl}methanesulfonamide (25). Following method B 4-methanesulfonamidoaniline (19) (0.49 g, 2.6 mM) yielded compound (25) (0.48 g, 62%), mp = 172–74 °C (dec.). IR (KBr): 3463, 3366, 3241, 2962, 2929, 2869, 1655, 1607, 1508, 1338, 1271, 1145, 1023, 767 cm−1. 1H-NMR (CDCl3): δ 7.56 (s, 1H), 7.31–7.28 (d, 2H, J = 6.7 Hz), 6.95 (s, 1H), 6.60–6.58 (d, 2H, J = 6.7 Hz), 4.96 (s, 2H), 4.17–4.13 (t, 1H, J = 7.5 Hz), 3.99 (s, 3H), 3.98 (s, 3H), 3.17 (s, 3H), 1.67–1.62 (m, 2H), 1.47–1.41 (m, 2H), 1.22–1.19 (m, 2H), 0.99–0.95 (t, 3H, J = 7.3 Hz). MS (EI) m/z: 460.1 [M]+. Anal. calcd for C22H28N4O5S: C, 57.37; H, 6.13; N, 12.17. Found: C, 57.28; H, 6.29; N, 12.34%.
4.1.10. 4-[(3-Butyl-3,4-dihydro-6,7-dimethoxy-4-oxoquinazolin-2-yl)methylamino]benzoic acid (26). Following method B 4-aminobenzoic acid (20) (0.36 g, 2.6 mM) afforded compound (26) (0.48 g, 69%), mp = 122–24 °C. IR (KBr): 3466, 3352, 3225, 2958, 2869, 1700, 1657, 1604, 1503, 1462, 1273, 1172, 1112, 1029, 772 cm−1. 1H-NMR (CDCl3): δ 7.91–7.89 (d, 2H, J = 8.6 Hz), 7.61 (s, 1H), 7.09 (s, 1H), 6.66–6.64 (d, 2H, J = 8.6 Hz), 5.37 (s, 2H), 4.14–4.08 (m, 1H), 4.01 (s, 3H), 3.99 (s, 3H), 1.78–1.74 (m, 2H), 1.42–1.36 (m, 2H), 0.91–0.88 (t, 3H, J = 7.5 Hz). MS (EI) m/z: 410.9 [M]+. Anal. calcd for C22H25N3O5: C, 64.22; H, 6.12; N, 10.21. Found: C, 64.51; H, 6.03; N, 10.13%.
4.1.11. 5-{4-[(3-Butyl-3,4-dihydro-6,7-dimethoxy-4-oxoquinazolin-2yl)methylamino]phenyl}-1H-tetrazole (27). Following method B 5-(4-aminophenyl)-1H-tetrazole (21) (0.39 g, 2.6 mM) afforded compound (27) (0.32 g, 44%), mp = 90–92 °C. IR (KBr): 3359, 3134, 2962, 1665, 1611, 1502, 1465, 1400, 1271, 1208, 1174, 1038 cm−1. 1H-NMR (DMSO-d6): δ 7.80–7.78 (d, 2H, J = 8.7), 7.49 (s, 1H), 6.90 (s, 3H), 6.72–6.69 (d, 2H, 1H, J = 8.7), 6.14 (s, 2H), 4.19–4.15 (t, 2H, J = 7.6 Hz), 3.98 (s, 3H), 3.93 (s, 3H), 1.63–1.55 (m, 2H), 1.48–1.40 (m, 2H), 0.99–0.94 (t, 3H, J = 7.6 Hz). MS (EI) m/z: 435.2 [M]+. Anal. calcd for C22H25N7O3: C, 60.68; H, 5.79; N, 22.51. Found: C, 60.46; H, 5.61; N, 22.73%.
4.1.12. 5-{3-[(3-Butyl-3,4-dihydro-6,7-dimethoxy-4-oxoquinazolin-2yl)methylamino]phenyl}-1H-tetrazole (28). Following method B 5-(3-aminophenyl)-1H-tetrazole (22) (0.39 g, 2.6 mM) afforded compound (28) (0.33 g, 46%), mp = 168–70 °C. IR (KBr): 3463, 3417, 3362, 3134, 2960, 1666, 1611, 1501, 1401, 1263, 1172, 1038, 788, 454, 420 cm−1. 1H-NMR (DMSO-d6): δ 7.78 (s, 1H), 7.52 (s, 1H), 7.46 (s, 1H), 7.41–7.39 (d, 1H, J = 7.6 Hz), 7.22–7.18 (m, 1H), 6.91 (s, 1H), 6.79–6.77 (d, 1H, J = 7.6 Hz), 6.14 (s, 2H), 4.66 (bs, 1H), 4.21–4.17 (t, 2H, J = 7.7 Hz), 3.95 (s, 3H), 3.92 (s, 3H), 1.67–1.59 (m, 2H), 1.51–1.43 (m, 2H), 0.88–0.85 (t, 3H). MS (EI) m/z: 435.0 [M]+. Anal. calcd for C22H25N7O3: C, 60.68; H, 5.79; N, 22.51. Found: C, 60.81; H, 5.64; N, 22.38%.
4.1.13. 2-[(4-Aminophenyl)aminomethyl]-3-butyl-6,7-dimethoxyquinazolin-4(3H)-one (29). Following method B p-phenylenediamine (23) (0.28 g, 2.6 mM) afforded compound (29) (0.52, 80%), mp = 136–38 °C. Rf 0.59 (chloroform:methanol, 9:1). IR (KBr): 3128, 2964, 1669, 1609, 1504, 1447, 1403, 1262, 1096, 1022, 802 cm−1. MS (EI) m/z: 381.2 [M]+. Anal. calcd for C21H26N4O3: C, 65.95; H, 6.85; N, 14.65. Found: C, 66.13; H, 6.59; N, 14.38%.
4.1.14. 2-{4-[(3-Butyl-3,4-dihydro-6,7-dimethoxy-4-oxoquinazolin-2-yl)methyl]piperazin-1-yl}-benzonitrile (33). Method C: 3-butyl-2-(chloromethyl)-6,7-dimethoxyquinazolin-4(3H)-one (17) (0.15 g, 0.48 mM), 1-(2-cyanophenyl)piperazine (30) (0.12 mL, 0.72 mM), flame dried potassium carbonate (0.28 g, 2.41 mM) and dry DMF (1 mL) were taken in an Rb flask (50 mL). The reaction mixture was stirred overnight at 60 °C and poured into ice-cold water (20 mL). The precipitate so obtained was filtered and washed several times with water. The solid thus obtained was recrystallized from DCM–methanol to afford compound (33) (0.15 g, 66%), mp = 171–73 °C. IR (KBr): 3414, 3068, 3003, 2961, 2828, 2220, 1670, 1593, 1497, 1448, 1402, 1266, 1237, 1054, 1016, 784 cm−1. 1H-NMR (DMSO-d6): δ 7.72–7.70 (d, 1H, J = 7.6 Hz), 7.62–7.58 (m, 1H), 7.43 (s, 1H), 7.17 (s, 1H), 7.14–7.08 (m, 2H), 4.16–4.13 (t, 3H, J = 7.6 Hz), 3.91 (s, 3H), 3.87 (s, 3H), 3.72 (s, 2H), 3.15 (bs, 4H), 2.67 (bs, 4H), 1.73–1.69 (m, 2H), 1.43–1.37 (m, 2H), 0.97–0.93 (t, 3H, J = 7.6 Hz). MS (EI) m/z: 460.8 [M]+. Anal. calcd for C26H31N5O3: C, 67.66; H, 6.77; N, 15.17. Found: C, 67.91; H, 6.52; N, 15.39%.
4.1.15. 3-Butyl-2-{[4-(2-fluorophenyl)piperazin-1-yl]methyl}-6,7-dimethoxyquinazolin-4(3H)-one (34). Following method C 1-(2-fluorophenyl)piperazine (31) (0.09 mL, 0.72 mM) yielded compound (34) (0.15 g, 68%), mp = 189–91 °C. IR (KBr): 2957, 2873, 2819, 1666, 1614, 1501, 1456, 1238, 1207, 1138, 1053, 1019, 757 cm−1. 1H-NMR (DMSO-d6): δ 7.43 (s, 1H), 7.16 (s, 1H), 7.13–7.08 (m, 2H), 7.03–6.96 (m, 2H), 4.16–4.12 (t, 2H, J = 7.9 Hz), 3.90 (s, 3H), 3.87 (s, 3H), 3.70 (s, 2H), 3.01 (bs, 4H), 2.65 (bs, 4H), 1.73–1.69 (m, 2H), 1.44–1.35 (m, 2H), 0.98–0.94 (t, 3H, J = 7.9 Hz). MS (EI) m/z: 453.9 [M]+. Anal. calcd for C25H31FN4O3: C, 66.06; H, 6.87; N, 12.33. Found: C, 65.87; H, 6.61; N, 11.96%.
4.1.16. 3-Butyl-6,7-dimethoxy-2-[(4-(pyridin-2-yl)piperazin-1-yl)methyl]quinazolin-4(3H)-one (35). Following method C 1-(2-pyridyl)piperazine (32) (0.11 mL, 0.72 mM) offered compound (35) (0.14 g, 64%), mp = 186–88 °C. IR (KBr): 3415, 3129, 3012, 2964, 2936, 2839, 2815, 1669, 1638, 1613, 1592, 1566, 1500, 1402, 1268, 1245, 1052, 1014, 784 cm−1. 1H-NMR (DMSO-d6): δ 8.11–8.10 (d, 1H, J = 7.5 Hz), 7.55–7.51 (m, 1H), 7.43 (s, 1H), 7.15 (s, 1H), 6.83–6.81 (d, 1H, J = 7.5 Hz), 6.66–6.63 (m, 1H), 4.17–4.13 (t, 2H, J = 7.7 Hz), 3.90 (s, 3H), 3.87 (s, 3H), 3.68 (s, 2H), 3.47 (bs, 4H), 2.58 (bs, 4H), 1.73–1.70 (m, 2H), 1.42–1.35 (m, 2H), 0.97–0.93 (t, 3H, J = 7.7 Hz). MS (EI) m/z: 437.0 [M]+. Anal. calcd for C24H31N5O3: C, 65.88; H, 7.14; N, 16.01. Found: C, 66.13; H, 7.09; N, 16.21%.
4.1.17. 2-(3-Aminophenylamino)-6,7-dimethoxyquinazolin-4-ylamine (38). Method D: to a solution of 4-amino-2-chloro-6,7-dimethoxyquinazoline (36) (0.2 g, 0.84 mM) in dry DMF, 3-phenylenediamine (37) (0.27 g, 2.51 mM) and flame dried potassium carbonate (0.23 g, 1.68 mM) were added. The reaction mixture was stirred overnight at 60 °C and poured into ice-cold water (20 mL). Precipitate so obtained was filtered and washed several times with water. Recrystallization from methanol–chloroform mixture afforded white colored compound (38) (0.15 g, 56%), mp = 245–47 °C. Rf 0.35 (20% methanol in chloroform). IR (KBr): 3460, 3369, 3321, 3198, 2935, 2839, 1647, 1626, 1576, 1500, 1439, 1315, 1236, 1215, 1011, 850 cm−1. 1H-NMR (DMSO-d6): δ 7.55 (s, 1H), 7.36 (s, 2H), 7.35 (s, 1H), 7.19 (bs, 1H), 7.04–6.97 (m, 2H), 6.42 (bs, 2H), 6.30–6.27 (d, 1H, J = 7.0 Hz), 3.98 (s, 2H), 3.95 (s, 3H), 3.94 (s, 3H). Anal. calcd for C16H17N5O2: C, 61.72; H, 5.50; N, 22.49. Found: C, 61.98; H, 5.37; N, 22.71%.
4.1.18. 2-[4-(4-Amino-6,7-dimethoxyquinazolin-2-yl)piperazin-1-yl]benzonitrile (42). Following method D 1-(2-cyanophenyl)piperazine (39) (0.43 mL, 2.50 mM) afforded compound (42) (0.17 g, 53%), mp = 127–29 °C. IR (KBr): 3491, 3321, 3198, 2930, 2837, 2219, 1639, 1571, 1519, 1485, 1440, 1379, 1284, 1253, 1218, 995, 850, 763 cm−1. 1H-NMR (DMSO-d6): δ 7.75–7.73 (d, 1H, J = 8.0 Hz), 7.64–7.60 (m, 1H), 7.43 (s, 1H), 7.23–7.21 (d, 1H, J = 8.0 Hz), 7.17 (bs, 2H), 7.14–7.06 (m, 1H), 6.75 (s, 1H), 3.90–3.89 (t, 4H), 3.84 (s, 3H), 3.79 (s, 3H), 3.19–3.17 (t, 4H, J = 4.4 Hz). MS (EI) m/z: 390.2 [M]+. Anal. calcd for C21H22N6O2: C, 64.60; H, 5.68; N, 21.52. Found: C, 64.93; H, 5.45; N, 21.69%.
4.1.19. 6,7-Dimethoxy-2-[4-(2-methoxyphenyl)piperazin-1-yl]quinazolin-4-ylamine (43). Following method D 1-(2-methoxyphenyl)piperazine (40) (0.45 mL, 2.50 mM) gave compound (43) (0.24 g, 73%), mp = 122–24 °C. IR (KBr): 3413, 3211, 2929, 2844, 1639, 1587, 1557, 1501, 1442, 1247, 1216, 1028, 751 cm−1. 1H-NMR (DMSO-d6): δ 7.42 (s, 1H), 7.13 (bs, 2H), 6.98–6.85 (m, 4H), 6.73 (s, 1H), 3.86 (bs, 3H), 3.83 (s, 3H), 3.81 (s, 3H), 3.79 (s, 3H), 2.99–2.96 (t, 4H, J = 4.8 Hz). MS (EI) m/z: 395.9 [M]+. Anal. calcd for C21H25N5O3: C, 63.78; H, 6.37; N, 17.71. Found: C, 64.05; H, 6.59; N, 17.39%.
4.1.20. 2-(4-Benzhydrylpiperazin-1-yl)-6,7-dimethoxyquinazolin-4-ylamine (44). Following method D benzhydrylpiperazine (41) (0.63 g, 2.50 mM) offered compound (44) (0.26 g, 68%), mp = 235–37 °C. IR (KBr): 3438, 3330, 3205, 3062, 2961, 2840, 1654, 1563, 1491, 1441, 1239, 1211, 1107, 998, 850, 749 cm−1. 1H-NMR (DMSO-d6): δ 7.46–7.45 (d, 4H, J = 7.2 Hz), 7.40 (s, 1H), 7.33–7.28 (m, 4H), 7.21–7.17 (m, 2H), 7.09 (bs, 2H), 6.68 (s, 1H), 4.30 (s, 1H), 3.80 (s, 3H), 3.77 (s, 3H), 3.71 (bs, 4H), 2.36–2.34 (t, 4H, J = 4.8 Hz). MS (EI) m/z: 456.3 [M]+. Anal. calcd for C27H29N5O2: C, 71.19; H, 6.42; N, 15.37. Found: C, 70.93; H, 6.58; N, 15.61%.
4.1.21. 6,7-Dimethoxy-2-(4-morpholinomethyl)quinazolin-4-ylamine (50). Method E: flame dried potassium carbonate (0.22 g, 1.58 mM) was added to a solution of 4-amino-2-chloromethyl-6,7-dimethoxyquinazoline (45) (0.2 g, 0.79 mM) and morpholine (46) (0.31 mL, 3.95 mM) in dry DMF. The reaction mixture was stirred overnight at 60 °C, poured into ice-cold water and the precipitate so obtained was filtered and washed with water to afford compound (50) (0.17 g, 72%), mp = 231–33 °C. IR (KBr): 3310, 3132, 3002, 2960, 2835, 1670, 1620, 1581, 1508, 1483, 1323, 1250, 1211, 1114, 993, 853 cm−1. 1H-NMR (DMSO-d6): δ 7.55 (s, 1H), 7.14 (s, 1H), 7.05 (bs, 2H), 3.95 (s, 6H), 3.71–3.69 (t, 4H, J = 4.5 Hz), 3.58 (s, 2H), 2.58 (bs, 4H). MS (EI) m/z: 305.0 [M]+. Anal. calcd for C15H20N4O3: C, 59.20; H, 6.62; N, 18.41. Found: C, 59.43; H, 6.46; N, 18.57%.
4.1.22. 6,7-Dimethoxy-2-[(piperidin-1-yl)methyl]quinazolin-4-ylamine (51). Following method E piperidine (47) (0.39 mL, 3.9 mM) afforded compound (51) (0.16 g, 67%), mp = 232–34 °C. Rf 0.16 (DCM:methanol, 19:1). IR (KBr): 3316, 3133, 3001, 2936, 2850, 1669, 1621, 1581, 1509, 1481, 1403, 1245, 1210, 983, 860, 782 cm−1. MS (EI) m/z: 303.0 [M]+. Anal. calcd for C16H22N4O2: C, 63.55; H, 7.33; N, 18.53. Found: C, 63.69; H, 7.05; N, 18.41%.
4.1.23. 2-[(4-Ethylpiperazin-1-yl)methyl]-6,7-dimethoxyquinazolin-4-ylamine (52). Following method E N-ethylpiperazine (48) (0.3 mL, 2.37 mM) offered compound (52) (0.17 g, 64%), mp = 241–43 °C. Rf 0.1 (DCM:methanol, 18:2). IR (KBr): 3307, 3147, 2945, 2819, 2699, 1666, 1620, 1580, 1491, 1280, 1248, 1132, 1016, 869 cm−1. MS (EI) m/z: 331.1 [M]+. Anal. calcd for C17H25N5O2: C, 61.61; H, 7.60; N, 21.13. Found: C, 61.78; H, 7.83; N, 21.29%.
4.1.24. 6,7-Dimethoxy-2-[(1H-1,2,4-triazol-1-yl)methyl]quinazolin-4-ylamine (53). Following method E 1,2,4-triazole (49) (0.27 g, 3.95 mM) afforded compound (53) (0.13 g, 56%), mp = 238–40 °C. IR (KBr): 3402, 3339, 3115, 2966, 2802, 1671, 1621, 1588, 1492, 1422, 1402, 1259, 1215, 1002, 771 cm−1. 1H-NMR (DMSO-d6): δ 8.52 (s, 1H), 7.89 (s, 1H), 7.63 (s, 1H), 7.11 (s, 1H), 5.46 (s, 2H), 3.97 (s, 3H), 3.96 (s, 3H). MS (EI) m/z: 286.0 [M]+. Anal. calcd for C13H14N6O2: C, 54.54; H, 4.93; N, 29.35. Found: C, 54.83; H, 5.21; N, 29.13%.
4.1.25. Methyl-3-[(4-amino-6,7-dimethoxyquinazolin-2-yl)methylamino]benzoate (61). 4-Amino-2-chloromethyl-6,7-dimethoxyquinazoline (45) (0.2 g, 0.79 mM) and methyl-3-aminobenzoate (54) (0.5 g, 3.95 mM) were reacted as per method E. The residue was recrystallized from DCM–methanol to afford the compound (61) (0.2 g, 69%), mp = 147–49 °C. IR (KBr): 3438, 3343, 3202, 2924, 2852, 1715, 1650, 1588, 1546, 1512, 1445, 1293, 1229, 1114, 999, 752 cm−1. 1H-NMR (DMSO-d6): δ 7.52 (s, 1H), 7.35 (bs, 2H), 7.03 (s, 1H), 7.00–6.96 (m, 1H), 6.60 (s, 1H), 6.55–6.52 (d, 1H, J = 8.0 Hz), 6.45–6.44 (d, 1H, J = 8.0 Hz), 6.09–6.08 (t, 1H, J = 4.6 Hz), 4.15–4.14 (d, 2H, J = 4.6 Hz), 3.87 (s, 3H), 3.86 (s, 3H), 3.84 (s, 3H). MS (EI) m/z: 368.4 [M]+. Anal. calcd for C19H20N4O4: C, 61.95; H, 5.47; N, 15.21. Found: C, 62.19; H, 5.63; N, 15.52%.
4.1.26. 3-[(4-Amino-6,7-dimethoxyquinazolin-2-yl)methylamino]benzoic acid (62). Following method E 3-aminobenzoic acid (0.54 g, 3.95 mM) afforded compound (62) (0.17 g, 62%), mp = 250–52 °C. IR (KBr): 3445, 3323, 3123, 1705, 1663, 1626, 1585, 1490, 1403, 1235, 1110, 982, 750 cm−1. 1H-NMR (DMSO-d6): δ 7.57 (s, 1H), 7.35 (s, 1H), 7.32 (bs, 2H), 7.29–7.27 (d, 1H, J = 7.8 Hz), 7.15–7.12 (m, 1H), 7.06 (s, 1H), 6.85–6.83 (d, 1H, J = 7.8 Hz), 5.23 (s, 2H), 4.93 (bs, 1H), 3.98 (s, 3H), 3.97 (s, 3H). MS (EI) m/z: 354.1 [M]+. Anal. calcd for C18H18N4O4: C, 61.01; H, 5.12; N, 15.81. Found: C, 60.83; H, 5.29; N, 15.74%.
4.1.27. 2-[(3-Toluidino)methyl]-6,7-dimethoxyquinazolin-4-ylamine (63). Following method E 3-toluidine (56) (0.54 g, 3.95 mM) offered compound (63) (0.16 g, 64%) mp = 160–62 °C. IR (KBr): 3477, 3396, 3301, 3119, 2830, 1644, 1615, 1578, 1506, 1403, 1213, 1169, 993, 767 cm−1. 1H-NMR (DMSO-d6): δ 7.52 (s, 1H), 7.29 (bs, 2H), 7.03 (s, 1H), 6.94–6.90 (m, 1H), 6.43 (s, 1H), 6.42–6.40 (d, 1H, J = 7.7 Hz), 6.35–6.33 (d, 1H, J = 7.7 Hz), 5.52 (bs, 1H), 4.14–4.13 (d, 2H, J = 4.3 Hz), 3.89 (s, 3H), 3.85 (s, 3H), 2.17 (s, 3H). Anal. calcd for C18H20N4O2: C, 66.65; H, 6.21; N, 17.27. Found: C, 66.41; H, 6.46; N, 17.51%.
4.1.28. 4-[(4-Amino-6,7-dimethoxyquinazolin-2-yl)methylamino]bromobenzene (64). Following method E 4-bromoaniline (57) (0.67 g, 3.95 mM) afforded compound (64) (0.18 g, 58%) mp = 217–19 °C. Rf 0.56 (DCM:methanol, 19:1). IR (KBr): 3381, 3342, 3128, 1673, 1622, 1586, 1494, 1371, 1240, 1207, 989, 857, 785 cm−1. MS (EI) m/z: 388 [M]+. Anal. calcd for C17H17BrN4O2: C, 52.46; H, 4.40; N, 14.39. Found: C, 52.71; H, 4.69; N, 14.45%.
4.1.29. 2-[(3-Chlorophenylamino)methyl]-6,7-dimethoxyquinazolin-4-ylamine (65). Following method E 3-chloroaniline (58) (0.41 mL, 3.95 mM) afforded compound (65) (0.17 g, 63%) mp = 219–21 °C. IR (KBr): 3503, 3388, 3321, 3126, 2958, 1658, 1621, 1583, 1510, 1483, 1258, 1211, 988, 819 cm−1. 1H-NMR (DMSO-d6): δ 8.39 (s, 1H), 8.09–8.07 (d, 1H, J = 8.0 Hz), 7.69–7.55 (m, 3H), 7.64 (s, 1H), 7.45–7.41 (m, 1H), 7.27 (s, 1H), 3.92 (s, 2H), 3.92 (s, 1H), 3.91 (s, 3H), 3.85 (s, 3H). MS (EI) m/z: 345 [M]+. Anal. calcd for C17H17ClN4O2: C, 59.22; H, 4.97; N, 16.25. Found: C, 58.95; H, 5.13; N, 16.12%.
4.1.30. 2-[(4-Chlorophenylamino)methyl]-6,7-dimethoxyquinazolin-4-ylamine (66). Following method E 4-chloroaniline (59) (0.5 g, 3.95 mM yielded compound (66)) (0.18 g, 66%) mp = 230–32 °C. IR (KBr): 3483, 3390, 3317, 3128, 2963, 2929, 1654, 1578, 1507, 1484, 1402, 1257, 1217, 1013, 816 cm−1. 1H-NMR (DMSO-d6): δ 7.52 (s, 1H), 7.29 (bs, 2H), 7.03 (s, 1H), 7.01–6.99 (d, 2H, J = 8.8 Hz), 6.61–6.58 (d, 2H, J = 8.8 Hz), 5.84 (bs, 1H), 4.14–4.13 (d, 2H, J = 3.7 Hz), 3.90 (s, 3H), 3.86 (s, 3H). MS (EI) m/z: 345 [M]+. Anal. calcd for C17H17ClN4O2: C, 59.22; H, 4.97; N, 16.25. Found: C, 59.03; H, 5.11; N, 16.18%.
4.1.31. 3-[(4-Amino-6,7-dimethoxyquinazolin-2-yl)methylamino]bromobenzene (67). Following method E 3-bromoaniline (60) (0.41 mL, 3.95 mM) offered compound (67) (0.184 g, 60%) mp = 234–36 °C. IR (KBr): 3422, 3122, 2930, 1656, 1611, 1502, 1401, 1345, 1003, 808 cm−1. 1H-NMR (DMSO-d6): δ 7.57 (s, 1H), 7.48 (bs, 2H), 7.07 (s, 1H), 7.01–6.97 (m, 1H), 6.80 (s, 1H), 6.65–6.62 (m, 2H), 6.40–6.38 (t, 1H, J = 5.3 Hz), 4.18–4.13 (d, 2H, J = 5.3 Hz), 3.88 (s, 3H), 3.85 (s, 3H). MS (EI) m/z: 389 [M]+. Anal. calcd for C17H17BrN4O2: C, 52.46; H, 4.40; N, 14.39. Found: C, 52.19; H, 4.63; N, 14.53%.
4.1.32. 6,7-Dimethoxy-2-[(4-phenylpiperazin-1-yl)methyl]quinazolin-4-ylamine (96). Following method E N-phenylpiperazine (68) (0.36 mL, 2.37 mM) afforded compound (96) (0.22 g, 74%) mp = 230–32 °C. IR (KBr): 3313, 3152, 3011, 2936, 2818, 1667, 1620, 1579, 1489, 1451, 1222, 1133, 1016, 773 cm−1. 1H-NMR (DMSO-d6): δ 7.55 (s, 1H), 7.44 (bs, 2H), 7.21–7.17 (m, 2H), 7.09 (s, 1H), 6.92–6.90 (d, 2H, J = 8.0 Hz), 6.77–6.74 (m, 1H), 3.89 (s, 3H), 3.85 (s, 3H), 3.49 (s, 2H), 3.11–3.09 (t, 4H, J = 4.6 Hz), 2.64–2.62 (t, 4H, J = 4.6 Hz). MS (EI) m/z: 379 [M]+. Anal. calcd for C21H25N5O2: C, 66.47; H, 6.64; N, 18.46. Found: C, 66.31; H, 6.87; N, 18.53%.
4.1.33. 6,7-Dimethoxy-2-[(4-(2-methoxyphenyl)piperazin-1-yl)methyl]quinazolin-4-ylamine (97). Following method E 1-(2-methoxyphenyl)piperazine (69) (0.41 mL, 1.58 mM) gave compound (97) (0.22 g, 71%) mp = 236–38 °C. IR (KBr): 3300, 3145, 3018, 2938, 2826, 1662, 1621, 1581, 1507, 1480, 1377, 1335, 1241, 1139, 1017, 791 cm−1. 1H-NMR (DMSO-d6): δ 7.89 (s, 1H), 7.59 (s, 1H), 7.18 (bs, 2H), 6.96–6.93 (d, 1H, J = 6.9 Hz), 6.91–6.85 (m, 3H), 3.96 (s, 6H), 3.83 (s, 3H), 3.68 (s, 2H), 3.11 (bs, 4H), 2.80 (bs, 4H). MS (EI) m/z: 409.0 [M]+. Anal. calcd for C22H27N5O3: C, 64.53; H, 6.65; N, 17.10. Found: C, 64.38; H, 6.56; N, 17.19%.
4.1.34. 4-Amino-2-[4-(benzoyl)piperazin-1-ylmethyl]-6,7-dimethoxyquinazoline (98). Following method E 1-benzoylpiperazine (70) (0.45 g, 2.37 mM), afforded compound (98) (0.32 g, 77%) as white solid, mp = 207–09 °C. IR (KBr): 3309, 3146, 2998, 2952, 2877, 2830, 1666, 1633, 1577, 1490, 1418, 1249, 1006, 868, 786 cm−1. 1H-NMR (CDCl3): δ 7.41–7.39 (m, 5H), 7.23 (s, 1H), 6.97 (s, 1H), 5.60 (bs, 2H), 4.00 (s, 3H), 3.97 (s, 3H), 3.87 (bs, 2H), 3.71 (s, 2H), 3.50 (bs, 2H), 2.69 (bs, 2H), 2.54 (bs, 2H). MS (ESI) m/z: 408.6 (M + 1)+. Anal. calcd for C22H25N5O3: C, 64.85; H, 6.18; N, 17.19. Found: C, 65.13; H, 5.89; N, 17.48%.
4.1.35. 4-Amino-2-[4-(2-fluorobenzoyl)piperazin-1-ylmethyl]-6,7-dimethoxyquinazoline (99). Following method E 1-(2-fluorobenzoyl)piperazine (71) (0.50 g, 2.37 mM), yielded compound (99) (0.34 g, 65%) as white solid, mp = 206–08 °C. IR (KBr): 3385, 3341, 3197, 2997, 2925, 2855, 1665, 1617, 1580, 1513, 1470, 1437, 1244, 1215, 1018, 866 cm−1. 1H-NMR (CDCl3): δ 7.39–7.34 (m, 2H), 7.22 (s, 1H), 7.20–7.16 (m, 1H), 7.09 (s, 1H), 7.07–7.05 (d, 1H, J = 8.7 Hz), 5.92 (bs, 2H), 3.99 (s, 3H), 3.94 (s, 3H), 3.88–3.87 (t, 2H, J = 4.5 Hz), 3.70 (s, 2H), 3.41 (bs, 2H), 2.70–2.68 (t, 2H, J = 4.5 Hz), 2.50–2.48 (bs, 2H). MS (ESI) m/z: 426.7 (M + 1)+. Anal. calcd for C22H24FN5O3: C, 62.11; H, 5.69; N, 16.46. Found: C, 61.83; H, 5.76; N, 16.71%.
4.1.36. 4-Amino-2-[4-(2-methoxybenzoyl)piperazin-1-ylmethyl]-6,7-dimethoxyquinazoline (100). Following method E 1-(2-methoxybenzoyl)piperazine (72) (0.52 g, 2.37 mM), offered compound (100) (0.35 g, 69%) as white solid, mp = 159–61 °C. IR (KBr): 3378, 3335, 3130, 2925, 2854, 1664, 1619, 1577, 1510, 1476, 1441, 1244, 1021, 755 cm−1. 1H-NMR (CDCl3): δ 7.34–7.22 (m, 2H), 7.21 (s, 1H), 7.10 (s, 1H), 6.97–6.94 (m, 1H), 6.88–6.86 (d, 1H, J = 7.8 Hz), 5.99 (bs, 2H), 3.99 (s, 3H), 3.95 (s, 3H), 3.89 (bs, 2H), 3.79 (s, 3H), 3.70 (s, 2H), 3.34 (bs, 2H), 2.65 (bs, 2H), 2.48 (bs, 2H). MS (ESI) m/z: 438.7 (M + 1)+. Anal. calcd for C23H27N5O4: C, 63.14; H, 6.22; N, 16.01. Found: C, 62.89; H, 6.29; N, 16.28%.
4.1.37. 4-Amino-2-[4-(3-methoxybenzoyl)piperazin-1-ylmethyl]-6,7-dimethoxyquinazoline (101). Following method E 1-(3-methoxybenzoyl)piperazine (73) (0.57 g, 5.41 mM), afforded compound (101) (0.30 g, 82%) as white solid. It was purified by column chromatography using methanol-chloroform (0.5%) as eluent, mp = 212–14 °C. IR (KBr): 3323, 3154, 3018, 2984, 2831, 1663, 1636, 1579, 1513, 1487, 1418, 1284, 1252, 987, 792 cm−1. 1H-NMR (CDCl3): δ 7.22–7.19 (m, 1H), 7.14 (s, 1H), 7.00 (s, 1H), 6.87–6.84 (m, 2H), 6.83 (s, 1H), 3.91 (s, 3H), 3.84 (s, 3H), 3.78 (bs, 2H), 3.72 (s, 3H), 3.63 (s, 2H), 3.42 (bs, 2H), 2.61 (bs, 2H), 2.46 (bs, 2H). MS (ESI) m/z: 437.8 (M + 1)+. Anal. calcd for C23H27N5O4: C, 63.14; H, 6.22; N, 16.01. Found: C, 63.02; H, 6.17; N, 16.19%.
4.1.38. 4-Amino-2-[4-(3-chlorobenzoyl)piperazin-1-ylmethyl]-6,7-dimethoxyquinazoline (102). Following method E 1-(3-chlorobenzoyl)piperazine (74) (0.74 g, 3.29 mM), yielded compound (102) (0.48 g, 72%) as white solid, mp = 225–27 °C. IR (KBr): 3320, 3150, 2954, 2831, 1663, 1638, 1578, 1512, 1488, 1402, 1238, 1212, 1008, 796 cm−1. 1H-NMR (CDCl3): δ 7.31 (s, 1H), 7.30–7.24 (m, 3H), 7.20 (s, 1H), 7.17 (s, 1H), 3.93 (s, 3H), 3.90 (s, 3H), 3.79 (bs, 2H), 3.65 (s, 2H), 3.42 (bs, 2H), 2.62 (bs, 2H), 2.49 (bs, 2H). MS (ESI) m/z: 442.8 (M + 1)+, 443.8 (M + 2)+. Anal. calcd for C22H24ClN5O4: C, 59.79; H, 5.47; N, 15.85. Found: C, 60.03; H, 5.51; N, 15.49%.
4.1.39. 4-Amino-2-[4-(2,3-dichlorobenzoyl)piperazin-1-ylmethyl]-6,7-dimethoxyquinazoline (103). Following method E 1-(2,3-dichlorobenzoyl)piperazine (75) (0.6 g, 2.37 mM), gave compound (103) (0.34 g, 67%) as white solid, mp = 259–61 °C (dec.). IR (KBr): 3347, 3140, 3002, 2958, 2830, 2803, 1675, 1620, 1578, 1513, 1488, 1438, 1243, 1211, 1008, 854 cm−1. 1H-NMR (CDCl3): δ 7.48–7.46 (d, 1H, J = 7.5 Hz), 7.25–7.23 (m, 2H), 7.20–7.18 (d, 1H, J = 7.5 Hz), 6.96 (s, 1H), 5.70 (bs, 2H), 4.00 (s, 3H), 3.98 (s, 3H), 3.93 (bs, 2H), 3.72 (s, 2H), 3.32 (bs, 2H), 2. 68 (bs, 2H), 2.51 (bs, 2H). 13C-NMR (DMSO-d6): 164.73, 160.83, 160.69, 153.84, 147.96, 137.78, 134.26, 132.35, 130.49, 127.59, 125.93, 115.32, 106.54, 102.65, 55.85, 55.47, 52.85, 52.37, 46.15. MS (ESI) m/z: 477.7 (M + 1)+, 478.7 (M + 2)+. Anal. calcd for C22H23Cl2N5O3: C, 55.47; H, 4.87; N, 14.70. Found: C, 55.68; H, 5.12; N, 14.63%.
4.1.40. 4-Amino-2-[4-(2,3-dimethoxybenzoyl)piperazin-1-ylmethyl]-6,7-dimethoxyquinazoline (104). Following method E 1-(2,3-dimethoxybenzoyl)piperazine (76) (0.59 g, 2.37 mM), afforded compound (104) (0.37 g, 67%) as white solid, mp = 145–47 °C. IR (KBr): 3344, 3212, 3001, 2936, 2835, 1621, 1581, 1510, 1480, 1437, 1267, 1000, 865, 783 cm−1. 1H-NMR (CDCl3): δ 7.21 (s, 1H), δ 7.09–7.05 (m, 1H), 7.02 (s, 1H), 6.92–6.90 (d, 1H, J = 7.7 Hz), 6.83–6.81 (d, 1H, J = 7.7 Hz), 5.75 (bs, 2H), 3.99 (s, 3H), 3.95 (s, 3H), 3.89 (s, 3H), 3.87 (s, 3H), 3.84 (s, 2H), 3.37 (bs, 2H), 2.8 (bs, 2H), 2.49 (bs, 2H), 2.48 (bs, 2H). 13C-NMR (CDCl3): 167.46, 161.17, 160.22, 154.95, 152.56, 149.03, 147.47, 144.96, 131.06, 124.73, 119.18, 116.47, 112.88, 107.44, 106.79, 100.41, 65.50, 61.51, 56.26, 56.20, 55.80, 53.81, 53.08, 46.82, 41.50. MS (ESI) m/z: 468.8 (M + 1)+. Anal. calcd for C24H29N5O5: C, 61.66; H, 6.25; N, 14.98. Found: C, 61.39; H, 6.39; N, 15.11%.
4.1.41. 4-Amino-2-[4-(3-methylbenzoyl)piperazin-1-ylmethyl]-6,7-dimethoxyquinazoline (105). Following method E 1-(3-methylbenzoyl)piperazine (77) (0.48 g, 2.37 mM), afforded compound (105) (0.22 g, 67%) as white solid, mp = 216–18 °C. IR (KBr): 3316, 3147, 3009, 2950, 2876, 2831, 1667, 1628, 1579, 1512, 1488, 1212, 1009, 868 cm−1. 1H-NMR (CDCl3): δ 7.24 (s, 1H), 7.21 (s, 1H), 7.19–7.06 (m, 3H), 6.91 (s, 1H), 5.62 (bs, 2H), 4.00 (s, 3H), 3.97 (s, 3H), 3.87 (bs, 2H), 3.71 (s, 2H), 3.50 (bs, 2H), 2.69 (bs, 2H), 2.62 (bs, 2H), 2.36 (s, 3H). MS (ESI) m/z: 422.9 (M + 1)+. Anal. calcd for C23H27N5O3: C, 65.54; H, 6.46; N, 16.62. Found: C, 65.38; H, 6.59; N, 16.70%.
4.1.42. 4-Amino-2-[4-(4-fluorobenzoyl)piperazin-1-ylmethyl]-6,7-dimethoxyquinazoline (106). Following method E 1-(4-fluorobenzoyl)piperazine (78) (0.47 g, 2.2 mM), gave compound (106) (0.32 g, 78%) as white solid, mp = 200–02 °C. IR (KBr): 3324, 3154, 2954, 2832, 1662, 1636, 1577, 1511, 1490, 1402, 1233, 1007, 846 cm−1. 1H-NMR (CDCl3): δ 7.43–7.40 (d, 2H, J = 8.5 Hz), 7.25 (s, 1H), 7.13–7.08 (d, 2H, J = 8.5), 7.06 (s, 1H), 6.30 (bs, 2H), 4.01 (s, 6H), 3.89 (bs, 2H), 3.74 (s, 2H), 3.55 (bs, 2H), 2.69 (bs, 2H), 2.59 (bs, 2H). MS (ESI) m/z: 426.7 (M + 1)+. Anal. calcd for C22H24FN5O3: C, 62.11; H, 5.69; N, 16.46. Found: C, 61.83; H, 6.01; N, 16.24%.
4.1.43. 4-Amino-2-[4-(4-cyanobenzoyl)piperazin-1-ylmethyl]-6,7-dimethoxyquinazoline (107). Following method E 1-(4-cyanobenzoyl)piperazine (79) (0.51 g, 2.37 mM), yielded compound (107) (0.34 g, 67%) as white solid, mp = 221–23 °C. IR (KBr): 3225, 3164, 3006, 2924, 2875, 2830, 2230, 1661, 1642, 1576, 1509, 1483, 1434, 1279, 1006, 848 cm−1. 1H-NMR (CDCl3): δ 7.71–7.69 (d, 2H, J = 7.5 Hz), 7.51–7.49 (d, 2H, J = 7.5 Hz), 7.22 (s, 1H), 6.99 (s, 1H), 4.00 (s, 3H), 3.95 (s, 3H), 3.87 (bs, 2H), 3.72 (s, 2H), 3.43 (bs, 2H), 2.71 (bs, 2H), 2.56 (bs, 2H). MS (ESI) m/z: 433.8 (M + 1)+. Anal. calcd for C23H24N6O3: C, 63.88; H, 5.59; N, 19.43. Found: C, 64.13; H, 5.29; N, 19.25%.
4.1.44. 4-Amino-2-[(4-benzylpiperazin-1-yl)methyl]-6,7-dimethoxyquinazoline (108). Following method E 1-benzylpiperazine (80) (0.72 g, 2.37 mM), yielded compound (108) (0.33 g, 72%) as white solid, mp = 206–08 °C. IR (KBr): 3429, 3133, 3028, 3005, 2972, 2872, 2812, 1629, 1589, 1514, 1400, 1281, 1139, 1012, 855, 749 cm−1. 1H-NMR (DMSO-d6): δ 7.76 (bs, 2H), 7.66 (s, 1H), 7.32–7.23 (m, 5H), 7.15 (s, 1H), 3.96 (s, 3H), 3.95 (s, 3H), 3.81 (bs, 4H), 3.73 (s, 2H), 3.58 (s, 2H), 2.81 (bs, 2H), 2.54 (bs, 2H). MS (ESI) m/z: 394.9 (M + 1)+. Anal. calcd for C22H27N5O2: C, 67.15; H, 6.92; N, 17.80. Found: C, 66.89; H, 7.15; N, 17.96%.
4.1.45. 4-Amino-2-[4-(2-trifluoromethylbenzyl)piperazin-1-ylmethyl]-6,7-dimethoxyquinazoline (109). Following method E 1-(2-trifluoromethylbenzyl)piperazine (81) (0.67 g, 2.37 mM), offered compound (109) (0.24 g, 67%) as white solid, mp = 221–23 °C. IR (KBr): 3497, 3430, 3290, 3063, 3028, 2943, 2877, 2805, 2766, 1641, 1577, 1510, 1476, 1274, 1060, 840, 774 cm−1. 1H-NMR (DMSO-d6): δ 7.79–7.77 (d, 1H, J = 7.7 Hz), 7.64–7.59 (m, 2H), 7.57 (s, 1H), 7.42–7.38 (d, 1H, J = 7.6 Hz), 7.37 (bs, 2H), 7.09 (s, 1H), 3.91 (s, 3H), 3.89 (s, 3H), 3.61 (s, 2H), 3.51 (s, 2H), 2.58 (bs, 4H), 2.46 (bs, 4H). 13C-NMR (DMSO-d6): 161.29, 160.67, 153.82, 147.85, 146.72, 137.39, 131.96, 130.14, 127.46, 126.83, 125.64, 125.29, 122.91, 106.58, 102.53, 65.17, 57.65, 55.82, 55.45, 52.96, 52.72. MS (ESI) m/z: 462.8 (M + 1)+. Anal. calcd for C23H26F3N5O2: C, 59.86; H, 5.68; N, 15.18. Found: C, 60.09; H, 5.83; N, 14.95%.
4.1.46. 4-Amino-2-[4-(2-methylbenzyl)piperazin-1-ylmethyl]-6,7-dimethoxyquinazoline (110). Following method E 1-(2-methylbenzyl)piperazine (82) (0.52 g, 2.37 mM), gave compound (110) (0.23 g, 71%) as white solid, mp = 223–25 °C. IR (KBr): 3491, 3071, 3002, 2958, 2933, 2801, 2759, 1634, 1563, 1506, 1479, 1435, 1246, 1009, 847, 755 cm−1. 1H-NMR (CDCl3): δ 7.26–7.24 (m, 4H), 7.14 (s, 1H), 6.88 (s, 1H), 5.44 (bs, 2H), 4.00 (s, 3H), 3.99 (s, 3H), 3.68 (s, 2H), 3.47 (s, 2H), 2.62 (bs, 4H), 2.56 (bs, 4H), 2.35 (s, 3H). 13C-NMR (CDCl3): 161.94, 160.13, 154.88, 148.94, 147.66, 137.48, 136.56, 130.18, 129.75, 126.90, 125.43, 107.66, 106.75, 100.04, 65.80, 60.76, 56.28, 56.18, 53.67, 52.99, 19.29. Anal. calcd for C23H29N5O2: C, 67.79; H, 7.17; N, 17.19. Found: C, 67.88; H, 6.91; N, 17.27%.
4.1.47. 4-Amino-2-[4-(2-bromobenzyl)piperazin-1-ylmethyl]-6,7-dimethoxyquinazoline (111). Following method E 1-(2-bromobenzyl)piperazine (83) (0.60 g, 2.37 mM), yielded compound (111) (0.27 g, 73%) as white solid, mp = 225–27 °C. IR (KBr): 3493, 3277, 3068, 3004, 2936, 2871, 2802, 2760, 1634, 1566, 1506, 1476, 1436, 1222, 1000, 756 cm−1. 1H-NMR (CDCl3): δ 7.53–7.51 (d, 1H, J = 7.8 Hz), 7.49–7.46 (d, 1H, J = 7.8 Hz), 7.28–7.26 (m, 1H), 7.24 (s, 1H), 7.11–7.06 (m, 1H), 6.91 (s, 1H), 5.69 (bs, 2H), 4.00 (s, 3H), 3.98 (s, 3H), 3.70 (s, 2H), 3.62 (s, 2H), 2.64 (bs, 8H). MS (ESI) m/z: 472.8 (M + 1)+, 473.8 (M + 2)+. Anal. calcd for C22H26BrN5O2: C, 55.94; H, 5.55; N, 14.83. Found: C, 56.13; H, 5.68; N, 15.05%.
4.1.48. 4-Amino-2-[4-(2-cyanobenzyl)piperazin-1-ylmethyl]-6,7-dimethoxyquinazoline (112). Following method E 1-(2-cyanobenzyl)piperazine (84) (0.55 g, 2.37 mM), offered compound (112) (0.29 g, 90%) as white solid, mp = 207–09 °C. IR (KBr): 3494, 3443, 3281, 3066, 2938, 2878, 2809, 2225, 1642, 1576, 1509, 1477, 1435, 1270, 1010, 866, 765 cm−1. 1H-NMR (CDCl3): δ 7.64–7.62 (d, 1H, J = 7.6 Hz), 7.56–7.53 (m, 2H), 7.35–7.31 (m, 1H), 7.27 (s, 1H), 7.24 (s, 1H), 5.58 (bs, 2H), 4.00 (s, 3H), 3.99 (s, 3H), 3.73 (s, 2H), 3.70 (s, 2H), 2.64 (bs, 8H). 13C-NMR (CDCl3): 161.74, 160.12, 154.91, 148.98, 147.62, 142.40, 132.88, 132.52, 130.05, 127.47, 117.89, 112.92, 107.62, 106.75, 100.09, 65.64, 60.37, 56.28, 56.22, 53.42, 52.68. MS (ESI) m/z: 419.9 (M + 1)+. Anal. calcd for C23H26N6O2: C, 66.01; H, 6.26; N, 20.08. Found: C, 66.29; H, 5.92; N, 20.13%.
4.1.49. 4-Amino-2-[4-(4-bromolbenzyl)piperazin-1-ylmethyl]-6,7-dimethoxyquinazoline (113). Following method E 1-(4-bromobenzyl)piperazine (85) (0.70 g, 2.37 mM), yielded compound (113) (0.23 g, 70%) as white solid, mp = 294–296 °C. IR (KBr): 3324, 3158, 3003, 2931, 2818, 1663, 1622, 1579, 1511, 1483, 1249, 1215, 1012, 867, 841 cm−1. 1H-NMR (CDCl3): δ 7.42–7.40 (d, 2H, J = 8.3 Hz), 7.23 (s, 1H), 7.19–7.17 (d, 2H, J = 8.3 Hz), 6.95 (s, 1H), 5.85 (bs, 2H), 3.98 (s, 3H), 3.94 (s, 3H), 3.68 (s, 2H), 3.44 (s, 2H), 2.63 (bs, 4H), 2.52 (bs, 4H). MS (ESI) m/z: 473.7 (M + 1)+, 474.7 (M + 2)+. Anal. calcd for C22H26BrN5O2: C, 55.94; H, 5.55; N, 14.83. Found: C, 56.19; H, 5.67; N, 15.07%.
4.1.50. 4-Amino-2-[4-(4-methylbenzyl)piperazin-1-ylmethyl]-6,7-dimethoxyquinazoline (114). Following method E 1-(4-methylbenzyl)piperazine (86) (0.52 g, 2.37 mM), afforded compound (114) (0.22 g, 69%) as white solid, mp = 229–31 °C. IR (KBr): 3313, 3148, 3007, 2933, 2825, 1663, 1619, 1578, 1512, 1486, 1014, 795 cm−1. 1H-NMR (CDCl3): δ 7.23 (s, 1H), 7.19–7.17 (d, 2H, J = 7.9 Hz), 7.11–7.09 (d, 2H, J = 7.9 Hz), 6.97 (s, 1H), 6.07 (bs, 2H), 3.98 (s, 3H), 3.93 (s, 3H), 3.68 (s, 2H), 3.47 (s, 2H), 2.63 (bs, 4H), 2.53 (bs, 4H), 2.32 (s, 3H). 13C-NMR (CDCl3): 161.85, 160.32, 154.83, 148.88, 147.59, 136.59, 134.89, 129.04, 128.85, 107.58, 106.78, 100.26, 65.73, 62.87, 56.24, 56.13, 53.47, 52.86, 21.10. MS (ESI) m/z: 408.6 (M + 1)+. Anal. calcd for C23H29N5O2: C, 67.79; H, 7.17; N, 17.19. Found: C, 68.02; H, 6.95; N, 17.35%.
4.1.51. 4-Amino-2-[4-(4-methoxybenzyl)piperazin-1-ylmethyl]-6,7-dimethoxyquinazoline (115). Following method E 1-(4-methoxybenzyl)piperazine (87) (0.57 g, 2.37 mM), offered compound (115) (0.24 g, 72%) as white solid, mp = 216–18 °C. IR (KBr): 3311, 3143, 3009, 2931, 2827, 1662, 1616, 1579, 1512, 1485, 1250, 1011, 844 cm−1. 1H-NMR (CDCl3): δ 7.24 (s, 1H), 7.23–7.21 (d, 2H, J = 9.0 Hz), 6.90 (s, 1H), 6.85–6.83 (d, 2H, J = 9.0 Hz), 5.63 (bs, 2H), 4.00 (s, 3H), 3.98 (s, 3H), 3.79 (s, 3H), 3.69 (s, 2H), 3.46 (s, 2H), 2.63 (bs, 4H), 2.54 (bs, 4H). MS (ESI) m/z: 424.8 (M + 1)+. Anal. calcd for C23H29N5O3: C, 65.23; H, 6.90; N, 16.54. Found: C, 65.39; H, 7.16; N, 16.83%.
4.1.52. 4-Amino-2-[4-(4-t-butylbenzyl)piperazin-1-ylmethyl]-6,7-dimethoxyquinazoline (116). Following method E 1-(4-t-butylbenzyl)piperazine (88) (0.63 g, 2.37 mM), gave compound (116) (0.28 g, 78%) as white solid, mp = 231–33 °C. IR (KBr): 3315, 3151, 3002, 2958, 2929, 2870, 2820, 1664, 1623, 1579, 1511, 1484, 1227, 1017, 848 cm−1. 1H-NMR (CDCl3): δ 7.32–7.30 (d, 2H, J = 8.2 Hz), 7.23 (s, 1H), 7.23–7.21 (d, 2H, J = 8.2 Hz), 6.93 (s, 1H), 5.85 (bs, 2H), 3.98 (s, 3H), 3.95 (s, 3H), 3.69 (s, 2H), 3.50 (s, 2H), 2.64 (bs, 4H), 2.55 (bs, 4H), 1.30 (s, 9H). 13C-NMR (CDCl3): 161.86, 160.10, 154.88, 149.90, 148.94, 147.66, 134.76, 129.09, 125.05, 107.67, 106.73, 100.02, 65.73, 62.71, 56.27, 56.19, 53.46, 52.74, 34.46, 31.40. Anal. calcd for C26H35N5O2: C, 69.46; H, 7.85; N, 15.58. Found: C, 69.13; H, 8.19; N, 15.85%.
4.1.53. 4-Amino-2-{4-[4-(2-cyanophenyl)benzyl]piperazin-1-ylmethyl}-6,7-dimethoxyquinazoline (117). Following method E 1-[4-(2-cyanophenyl)benzyl]piperazine (89) (0.76 g, 2.37 mM), yielded compound (117) (0.31 g, 79%) as white solid, mp = 230–32 °C. IR (KBr): 3301, 3133, 3004, 2929, 2829, 2807, 2224, 1663, 1619, 1578, 1557, 1512, 1489, 1142, 1012, 869, 764 cm−1. 1H-NMR (DMSO-d6): δ 7.84–7.82 (d, 1H, J = 7.8 Hz), 7.75–7.71 (m, 1H), 7.57 (s, 1H), 7.55–7.54 (m, 2H), 7.52–7.50 (d, 2H, J = 8.2 Hz), 7.45–7.43 (d, 2H, J = 8.2 Hz), 7.25 (bs, 2H), 7.09 (s, 1H), 3.91 (s, 6H), 3.56 (s, 2H), 3.53 (s, 2H), 2.59 (bs, 4H), 2.49 (bs, 4H). MS (ESI) m/z: 495.9 (M + 1)+. Anal. calcd for C29H30N6O2: C, 70.42; H, 6.11; N, 16.99. Found: C, 70.57; H, 6.19; N, 17.08%.
4.1.54. 4-Amino-2-[4-(1-naphthylmethyl)piperazin-1-ylmethyl]-6,7-dimethoxyquinazoline (118). Following method E 1-(1-naphthylmethyl)piperazine (90) (0.62 g, 2.37 mM), offered compound (118) (0.24 g, 69%) as white solid, mp = 217–19 °C. IR (KBr): 3431, 3306, 3156, 3006, 2935, 2873, 2809, 2767, 1633, 1582, 1512, 1478, 1137, 1012, 843, 784 cm−1. 1H-NMR (DMSO-d6): δ 8.26–8.24 (d, 1H, J = 7.5 Hz), 7.87–7.85 (d, 1H, J = 7.5 Hz), 7.80–7.78 (m, 1H), 7.56 (s, 1H), 7.53–7.50 (m, 2H), 7.48–7.46 (d, 2H), 7.42 (bs, 2H), 7.09 (s, 1H), 3.90 (s, 3H), 3.89 (s, 3H), 3.88 (s, 2H), 3.55 (s, 2H), 2.60 (bs, 4H), 2.53 (bs, 4H). 13C-NMR (DMSO-d6): 160.69, 153.92, 147.94, 133.65, 133.33, 132.00, 128.00, 127.54, 127.14, 125.51, 125.39, 124.92, 124.58, 106.51, 106.31, 102.60, 99.49, 60.45, 55.87, 55.49, 52.91, 52.54. MS (ESI) m/z: 444.7 (M + 1)+. Anal. calcd for C26H39N5O2: C, 70.41; H, 6.59; N, 15.79. Found: C, 70.73; H, 6.68; N, 15.85%.
4.1.55. 4-Amino-2-[(4-benzhydryl)piperazin-1-ylmethyl]-6,7-dimethoxyquinazoline (119). Following method E 1-benzhyrylpiperazine (91) (0.79 g, 3.16 mM), afforded compound (119) (0.29 g, 75%) as white solid, mp = 234–36 °C. IR (KBr): 3491, 3026, 2972, 2943, 2872, 2759, 1647, 1578, 1564, 1509, 1481, 1250, 1007, 851, 706 cm−1. 1H-NMR (CDCl3): δ 7.41–7.39 (d, 4H, J = 7.4 Hz), 7.26 (s, 1H), 7.26–7.21 (m, 4H), 7.16–7.13 (m, 2H), 6.92 (s, 1H), 5.71 (bs, 2H), 4.23 (s, 1H), 3.98 (s, 3H), 3.95 (s, 3H), 3.70 (s, 2H), 2.65 (bs, 4H), 2.49 (bs, 4H). 13C NMR (CDCl3): 161.71, 160.13, 154.88, 148.93, 147.58, 142.80, 128.41, 127.95, 126.85, 107.59, 106.73, 100.14, 65.63, 56.26, 56.19, 53.79, 51.61. Anal. calcd for C28H31N5O2: C, 71.62; H, 6.65; N, 14.91. Found: C, 71.85; H, 6.73; N, 15.15%.
4.1.56. 4-Amino-2-[4-(cyclohexylmethyl)piperazin-1-ylmethyl]-6,7-dimethoxyquinazoline (120). Following method E 1-(cyclohexylmethyl)piperazine (92) (0.57 g, 2.37 mM), yielded compound (120) (0.21 g, 66%) as white solid, mp = 206–08 °C. IR (KBr): 3338, 3156, 3001, 2924, 2846, 1660, 1623, 1580, 1512, 1489, 1255, 1227, 1019, 868 cm−1. 1H-NMR (CDCl3): δ 7.24 (s, 1H), 6.92 (s, 1H), 5.72 (bs, 2H), 4.00 (s, 3H), 3.98 (s, 3H), 3.69 (s, 2H), 2.65 (bs, 4H), 2.51 (bs, 4H), 2.13 (s, 2H), 1.77–1.64 (m, 4H), 1.50–1.45 (m, 1H), 1.26–1.13 (m, 4H), 0.90–0.81 (m, 2H). Anal. calcd for C22H33N5O2: C, 66.14; H, 8.33; N, 17.53. Found: C, 65.85; H, 8.52; N, 17.78%.
4.1.57. 1-[4-(4-Amino-6,7-dimethoxyquinazolin-2-yl)methylpiperazin-1-yl]-2-phenylethanone (121). Following method E 2-phenyl-1-(piperazin-1-yl)ethanone (93) (0.83 g, 2.96 mM), offered compound (121) (0.42 g, 88%) as white solid, mp = 219–21 °C. IR (KBr): 3336, 3164, 2965, 2832, 1657, 1618, 1577, 1553, 1511, 1400, 1255, 1227, 988, 868 cm−1. 1H-NMR (CDCl3): δ 7.25 (bs, 2H), 7.23 (s, 1H), 7.21–7.20 (d, 2H), 7.15–7.14 (m, 3H), 6.88 (s, 1H), 3.93 (s, 3H), 3.91 (s, 3H), 3.66 (bs, 2H), 3.66 (s, 2H), 3.57 (s, 2H), 3.47–3.45 (t, 2H, J = 4.5 Hz), 2.50–2.48 (t, 2H, J = 4.5 Hz), 2.34 (t, 2H). MS (ESI) m/z: 422.9 (M + 1)+. Anal. calcd for C23H27N5O3: C, 65.54; H, 6.46; N, 16.62. Found: C, 65.83; H, 6.59; N, 16.49%.
4.1.58. 1-[4-(4-Amino-6,7-dimethoxyquinazolin-2-yl)methylpiperazin-1-yl]-2-(3-chlorophenyl)ethanone (122). Following method E 2-(3-chlorophenyl)-1-(piperazin-1-yl)ethanone (94) (0.56 g, 2.37 mM), yielded compound (122) (0.42 g, 75%) as white solid, mp = 220–22 °C. IR (KBr): 3332, 3146, 3004, 2962, 2829, 1657, 1618, 1577, 1554, 1511, 1488, 1254, 1227, 987, 866, 764 cm−1. 1H-NMR (CDCl3): δ 7.24 (s, 1H), 7.23 (s, 1H), 7.22–7.21 (d, 2H), 7.13–7.11 (m, 1H), 6.92 (s, 1H), 4.01 (s, 3H), 4.00 (s, 3H), 3.75–3.73 (t, 2H, J = 5.0 Hz), 3.69 (s, 2H), 3.67 (s, 2H), 3.54–3.51 (t, 2H, J = 5.0 Hz), 2.59–2.56 (t, 2H, J = 5.0 Hz), 2.48–2.45 (t, 2H, J = 5.0 Hz). MS (ESI) m/z: 456.7 (M + 1)+, 457.7 (M + 2)+. Anal. calcd for C23H26ClN5O3: C, 60.59; H, 5.75; N, 15.36. Found: C, 60.71; H, 5.48; N, 15.51%.
4.1.59. 1-[4-(4-Amino-6,7-dimethoxyquinazolin-2-yl)methylpiperazin-1-yl]-2-(4-methoxyphenyl)ethanone (123). Following method E 2-(4-methoxyphenyl)-1-(piperazin-1-yl)ethanone (95) (0.55 g, 2.37 mM), offered compound (123) (0.26 g, 63%) as white solid, mp = 212–14 °C. IR (KBr): 3333, 3161, 3009, 2960, 2833, 1657, 1616, 1578, 1512, 1490, 1252, 1226, 988, 865, 788 cm−1. 1H-NMR (CDCl3): δ 7.23 (s, 1H), 7.15–7.13 (d, 2H, J = 8.9 Hz), 6.96 (s, 1H), 6.86–6.82 (d, 2H, J = 8.9 Hz), 4.01 (s, 3H), 4.00 (s, 3H), 3.79 (s, 3H), 3.75–3.72 (t, 2H, J = 4.9 Hz), 3.66 (s, 2H), 3.65 (s, 2H), 3.55–3.53 (t, 2H, J = 4.9 Hz), 2.57–2.55 (t, 2H, J = 4.9 Hz), 2.44–2.41 (t, 2H, J = 4.9 Hz). MS (ESI) m/z: 452.9 (M + 1)+. Anal. calcd for C24H29N5O4: C, 63.84; H, 6.47; N, 15.51. Found: C, 64.13; H, 6.53; N, 15.44%.
4.2. Biological studies
All the experiments were performed on healthy adult male Wistar rats (300–330 g) in accordance with guidelines provided by the committee for the purpose of control and supervision of experiments on animals (CPCSEA), New Delhi, India. All the experimental protocols were approved by the institutional animals ethics committee (IAEC) of Faculty of Pharmacy, The M. S. University of Baroda (approval no. MSU/IAEC/2014-15/1435).
4.2.1. Functional antagonism assay on rat aortic strip. Male Wistar rats were sacrificed humanely. Descending segment of thoracic aortas were dissected free immediately and placed in ice-cold and aerated Kreb's bicarbonate solution of the following composition (mM): NaCl 112, NaHCO3 12, glucose 11.1, KCl 5.0, MgSO4 1.2, KH2PO4 1.0 and CaCl2 2.5.33 Adherent fat was removed carefully and gentle rubbing was applied to denude endothelium. Following this, aortic strip was prepared using surgical blade and both ends of the strips were tied by a thread. The strips were suspended in prewarmed oxygenated Kreb's solution under the resting tension of 2 g during experiment. Before performing of the experiment, the strip was allowed to stabilize for 60 minutes during which Kreb's solution was changed for every 10 minutes. Removal of the endothelium was confirmed by observing the “absence of relaxation” on strips precontracted with potassium chloride/phenylephrine. Isotonic contractions were recorded using a transducer (UGO BASILE, Italy) coupled to a Gemini 7070 recorder (UGO BASILE, Italy). Graded and cumulative contractions were induced by phenylephrine and AII separately and dose response curves were obtained. Frequent washing was performed by Kreb's solution and the strip was allowed to relax completely. Test compounds/standard antagonists were then added to the organ tubes and incubated for 30 minutes prior to the addition of phenylephrine/AII. Control strips were incubated with solvent [DMSO (0.5%) or normal saline] for 30 minutes prior to recording the concentration response curves. Antagonism of test compounds were determined by finding out pA2 values of the test compounds.34
4.2.2. Invasive measurement of blood pressure. The effects of compounds (42 and 110) were found to have considerable antagonistic activity on both the receptors in the in vitro isolated tissue studies, so it was decided to challenge the in vivo effects of phenylephrine and AII on rat arterial blood pressure by these compounds (42 and 110) independently. This method involves measurement of arterial blood pressure via catheterization of the carotid artery and provides a direct measurement of blood pressure.35 Briefly, after anesthetizing the animals, partial cut was put on trachea that was cannulised for allowing unobstructed respiration. Vagotomy was performed to exclude the effect of vagus nerve on presser response. The left femoral vein was cannulated for administration of test substances. The carotid artery was isolated and cannulated with a polyethylene catheter attached to a pressure transducer (MLT 844 pressure transducer, Power Lab) for measurement of intra-arterial blood pressure. The transducer was filled with heparinized saline (100 IU per mL) to prevent blood clotting in the transducer and connected to the Powerlab-4/35 data acquisition system (AD Instruments, Australia) for recording of the blood pressure. Following this, the animal was allowed to stabilize for 30 to 45 minutes to remove surgical stress. Baseline blood pressure was recorded following which responses to the agonists (in absence and presence of the antagonists) were observed.
4.3. Molecular modeling studies
4.3.1. Pharmacophore and 3D-QSAR. To develop a pharmacophore model and on its basis a 3D-QSAR model, the compounds reported19 previously from this lab as well as the newly synthesized ones were used. To develop the pharmacophore and 3D-QSAR models, the PHASE module of Schrodinger was utilized.36 PHASE works on tree-based partitioning algorithm and systematically identifies the spatial arrangements of functional groups essential for the biological activity from a set of high-affinity ligands. Along with the activity data, the obtained pharmacophore hypothesis offers a 3D-QSAR model to recognize the structural features which govern the activity. In order to develop pharmacophore and 3D-QSAR models, structures of the ligands (test compounds) were built within Maestro and prepared using LigPrep module of the Schrodinger at physiological pH. For the development of both the α1 and AT1 pharmacophore models, conformations for all the ligands under study were searched by combination of Monte-Carlo multiple minimum (MCMM)/Low mode (LMOD) with a maximum number of 250 conformers per ligand and 100 steps of minimization. A pharm-set was assigned for the development of α1 model with pA2 value threshold of 8 and above as actives and pA2 value 5 and below as inactive. Same parameters for actives and inactive were used for developing a model for AT1-antagonists as used for α1-antagonists.In order to validate the developed 3D-QSAR models, extensive validation study was performed by using various validation parameters. The k and k′ values were determined by using regression line with no intercept by exchanging X and Y axis.37,38 Further, statistical techniques described by Saha and Raghava were also followed.39 Here, a sensitivity value indicates that fraction of the compounds which are properly predicted as ‘actives’ from the total active molecules, a specificity value indicates proportion of the molecules which are appropriately predicted as ‘non-actives’ out of the total non-active compounds, an accuracy value was determined to identify whether the compounds are correctly divided and predicted as true actives or true inactives, a positive prediction value (PPV) or precision, a negative prediction value (NPV), and a Matthew's correlation coefficient (MCC) value were also determined for the developed 3D-QSAR models. To perform the statistical parameters proposed by Saha and Raghava accurately, as verified by our group previously,40,41 the data set has to be divided in approximately equal halves and one part has to be considered as active and the other as inactive to identify true positives, true negatives, false positives and false negatives. Here, in the case of α1 model the threshold value was 6.5 and for AT1 model it was considered at 6.3. Further the condition of {[(R2 − R02)/R2] < 0.1 or [(R2 − R0′2)/R2] < 0.1} was also implemented.37 Further, to validate the coefficient of determination values the modified test i.e. Rm2 test was implemented.42
4.3.2. Homology modeling and docking studies. To understand the molecular mechanism of dual acting anti-hypertensive agents with α1 and AT1 receptors antagonism, docking studies of the synthesized compounds were performed within the active sites of the respective α1- and AT1-receptors obtained by homology modeling. Both the receptors belong to the family of GPCRs. To derive a homology model of α1-adrenergic receptor, the amino acid sequence was obtained from Swiss-Prot database (entry id: P35348). The crystal structure of Bovine Rhodopsin (PDB Code: 1F88; resolution 2.80 Å) was used as template to generate homologous model of α1-adrenergic receptor. For the development of a homology model of α1-receptor, the sequences under study were aligned using clustalW multiple sequence alignment and the initial 21 amino acid sequence of α1 was truncated and the model was developed. As both the receptors under study are GPCRs, the 3D structure of AT1 was developed similarly using the bovine rhodopsin as template in MODELLER.43 The amino acid sequence of AT1 was obtained from Swiss-Prot database (entry id: P30556).The 3D models for the α1- and AT1-receptors were developed by means of MODELLER. The stereochemical qualities of the developed and minimized structures were evaluated with the help of PROCHECK44 software which suggested a high quality of the developed models with a commendable distribution of ψ and φ angels in Ramachandran plots. After optimization, the generated 3D structures of the receptors were prepared with the aid of protein preparation wizard within Schrodinger.
In order to evaluate the ligand receptor interactions, docking studies were performed for compounds (110 and 3) in the α1 series, on the active site of α1-adrenergic receptor. Whereas compounds (110 and 130) for AT1-antagonist activity from the series under study were checked by docking these compounds in the active site of AT1-receptor. The docking studies were performed by using Glide module with XP mode.45 The ligand structures were generated within Maestro and prepared using LigPrep module of Schrodinger.42
Acknowledgements
Prof. M. R. Yadav is thankful to UGC, New Delhi for the award of ‘UGC-BSR One time research grant’ [F.19-147/2015(BSR)]. The study was also supported by UGC-BSR-RFMS fellowship to NA, AMK and SP. JM, thanks UGC project (F. No. 41-716/2012-SR). The authors acknowledge the analytical facilities provided by Dr Vikram Sarabhai Research Center, The M. S. University of Baroda, Vadodara, India.
References
- http://www.who.int/topics/hypertension/en/, accessed on 27.03.2015.
- M. Moser, Adv. Intern. Med., 2001, 161, 1140–1144 CAS.
- G. Mancia, Eur. Heart J., 2007, 28, 1462–1536 Search PubMed.
- P. Allegretti, S. Ki Choi, R. Gendron, P. R. Fatheree and R. M. McKinnell, US Pat., US 8501798B2, 2013.
- P. Allegretti, S. Ki Choi, R. Gendron, P. R. Fatheree and R. M. McKinnell, US Pat., US 20080269305A1, 2008.
- R. Morphy and Z. Rancovic, Curr. Pharm. Des., 2009, 15, 587–600 CrossRef CAS PubMed.
- C. G. Wermuth, Drug Discovery Today, 2004, 9, 826–827 CrossRef PubMed.
- S. Rizzo, M. Bartolini, L. Ceccarini, L. Piazzi, S. Gobbi, A. Cavalli, M. Recanatini, V. Andrisano and A. Rampa, Bioorg. Med. Chem., 2010, 18, 1749–1760 CrossRef CAS PubMed.
- P. Zhan and X. Liu, Curr. Pharm. Des., 2009, 15, 1893–1917 CrossRef CAS PubMed.
- R. Morphy and Z. Rankovic, Drug Discovery Today, 2007, 12, 156–159 CrossRef CAS PubMed.
- Z. Haliin, F. Mati and Y. Moussa, Pharmaceuticals, 2014, 7, 113–135 CrossRef PubMed.
- P. Csermely, V. Agoston and S. Pongor, Trends Pharmacol. Sci., 2005, 26, 178–182 CrossRef CAS PubMed.
- T. F. Walsh, K. J. Fitch, D. L. Williams, K. L. Murphy, N. A. Nolan, D. J. Pettibone, R. S. L. Chang, S. S. Malley, B. V. Clineschmidt, D. F. Veber and W. J. Greenlee, Bioorg. Med. Chem. Lett., 1995, 5, 1155–1158 CrossRef CAS.
- N. Murugesan, Z. X. Gu, L. Fadnis, J. E. Tellew, R. A. F. Baska, Y. F. Yang, S. M. Beyer, H. Monshizadegan, K. E. Dickinson, M. T. Valentine, H. W. Griffith, S. J. Lan, W. R. Ewing, K. E. Carlson, M. C. Kowala, R. Zahler and J. E. Maco, J. Med. Chem., 2005, 48, 171–179 CrossRef CAS PubMed.
- N. Murugesan, J. E. Tellew, Z. X. Gu, B. L. Kunst, L. Fadnis, L. A. Cornelius, R. A. F. Baska, Y. F. Yang, S. M. Beyer, H. Monshizadegan, K. E. Dickinson, B. Panchal, M. T. Valentine, S. Chong, R. A. Morrison, K. E. Carlson, J. R. Powell, S. Moreland, J. C. Barrish, M. C. Kowala and J. E. Macor, J. Med. Chem., 2002, 45, 3829–3835 CrossRef CAS PubMed.
- B. N. C. Prichard and B. Tomlinson, Drugs, 1988, 36, 20–25 CrossRef PubMed.
- M. Yanagisawa, H. Kurihara, S. Kimura, Y. Tomobe and M. Kobayashi, Nature, 1988, 332, 411–415 CrossRef CAS PubMed.
- P. Naik, P. Murumkar, R. Giridhar and M. R. Yadav, Bioorg. Med. Chem., 2010, 18, 8418–8456 CrossRef CAS PubMed.
- M. R. Yadav, P. P. Naik, H. P. Gandhi, B. S. Chauhan and R. Giridhar, Bioorg. Med. Chem. Lett., 2013, 23, 3959–3966 CrossRef CAS PubMed.
- A. Bali, K. Sharma, A. Bhalla, S. Bala and D. Reddy, Eur. J. Med. Chem., 2010, 45, 2656–2662 CrossRef CAS PubMed.
- N. Murugesan, Z. Gu, L. Fadnis, J. E. Tellew, R. A. Baska, Y. Yang and S. M. Beyer, J. Med. Chem., 2005, 13, 171–179 CrossRef PubMed.
- A. B. Pinkerton, R. Ardecky, E. A. Serguienko, M. Gonzalez-lopez, S. R. Ganji and J. Zou, US Pat., US 048570A2, 2015.
- L. X. Wang, X. B. Zhou, M. L. Xiao, N. Jiang, F. Liu, W. X. Zhou, X. K. Wanga, Z. B. Zheng and S. Li, Bioorg. Med. Chem. Lett., 2014, 24, 3739–3743 CrossRef CAS PubMed.
- S. K. Verma, R. Ghorpade, A. Pratap and M. P. Kaushik, Green Chem., 2012, 14, 326–329 RSC.
- C. Zhang, C. Tan, X. Zu, X. F. Liu, B. Chu, X. Ma, Y. Chen, P. Gong and Y. Jiang, Eur. J. Med. Chem., 2011, 45, 1404–1414 CrossRef PubMed.
- D. C. Hsu, H. S. Roth, D. C. West, R. C. Botham, C. J. Novotny, S. C. Schmid and P. J. Hergenrother, ACS Comb. Sci., 2012, 14, 44–50 CrossRef CAS PubMed.
- G. Y. Meti, R. R. Kamble, D. B. Biradar and S. B. Margankop, Med. Chem. Res., 2013, 22, 5868–5877 CrossRef CAS.
- S. Parsuraman and R. Raveendran, J. Pharmacol. Pharmacother., 2012, 3, 172–177 Search PubMed.
- F. Triposkiadis, G. Karayannis, G. Giamouzis, J. Skoularigis, G. Louridas and J. Butler, J. Am. Coll. Cardiol., 2009, 54, 1747–1762 CrossRef CAS PubMed.
- J. T. Liles, P. A. Dabisch and K. E. Hude, J. Pharmacol. Exp. Ther., 2006, 316, 95–105 CrossRef CAS PubMed.
- S. H. Padia, N. L. Howell and H. M. Siragy, Hypertension, 2006, 47, 537–544 CrossRef CAS PubMed.
- M. R. Yadav, H. P. Gandhi, P. P. Naik and R. Giridhar, Pharm. Biol., 2012, 50, 439–442 CrossRef CAS PubMed.
- M. Clozel, C. Binkert, M. Birker-Robaczewska, C. Boukhadra, S. S. Ding and W. Fischli, J. Pharmacol. Exp. Ther., 2004, 311, 204–212 CrossRef CAS PubMed.
- O. Arunlakshana and H. O. Schild, Br. J. Pharmacol. Chemother., 1959, 14, 48–58 CrossRef CAS PubMed.
- T. W. Kurtz, K. A. Griffin and A. K. Bidani, Hypertension, 2005, 45, 299–310 CrossRef CAS PubMed.
- S. L. Dixon, A. M. Smondyrev and S. N. Rao, Chem. Biol. Drug Des., 2006, 67, 370–372 CAS.
- A. Golbraikh and A. Tropsha, J. Mol. Graphics Modell., 2002, 20, 269–276 CrossRef CAS PubMed.
- A. Golbraikh and A. Tropsha, J. Comput.-Aided Mol. Des., 2002, 16, 357–369 CrossRef CAS PubMed.
- S. Saha and G. Raghava, Nucleic Acids Res., 2006, 34, 202–209 CrossRef PubMed.
- A. M. Kanhed, V. P. Zambre, V. A. Pawar, M. K. Sharma, R. Giridhar and M. R. Yadav, Med. Chem. Res., 2014, 23, 5215–5223 CrossRef CAS.
- A. M. Kanhed, R. C. Dash, N. Parmar, T. Kumar, R. Giridhar and M. R. Yadav, Mol. Diversity, 2015, 19, 965–974 CrossRef CAS PubMed.
- P. P. Roy, S. M. Paul and K. Roy, Molecules, 2009, 14, 1660–1701 CrossRef CAS PubMed.
- M. A. Marti-Renom, A. C. Stuart, A. Fiser, R. Sanchez, F. Melo and A. Sali, Annu. Rev. Biophys. Biomol. Struct., 2000, 29, 291–325 CrossRef CAS PubMed.
- L. Stahle and S. Wold, J. Chemom., 1987, 1, 185–196 CrossRef.
- T. A. Halgren, R. B. Murphy, R. A. Friesner, H. S. Beard, L. L. Frye, W. T. Pollard and J. L. Banks, J. Med. Chem., 2004, 47, 1750–1759 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra00589f |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.