
Nanoparticles and mesenchymal stem cells: a win-win alliance for anticancer drug delivery
Min Li,
Fangrong Zhang,
Kerong Chen,
Cheng Wang,
Yujie Su,
Yuan Liu,
Jianping Zhou* and
Wei Wang*
State Key Laboratory of Natural Medicines, Department of Pharmaceutics, China Pharmaceutical University, 24 Tongjiaxiang, Nanjing 210009, China. E-mail: wangcpu209@cpu.edu.cn; zhoujp60@163.com
First published on 7th April 2016
Abstract
The development of nanoparticles (NPs) provides anticancer therapy with new strategies, such as noninvasive imaging and targeted drug delivery. However, uneven intratumoral distribution and immunogenicity hinder the clinical applications of NPs. Currently, mesenchymal stem cells (MSCs) are emerging as a hotspot in anticancer drug delivery due to their intrinsic characteristics, such as tumor-tropism and low immunogenicity. Herein, the combination of MSCs and NPs offers an advanced option to overcome the limitations of NPs. On the other hand, NPs could benefit MSC-based drug delivery systems in many aspects. Firstly, genetic modification is currently used in the design of cell vectors. More and more researchers prefer to use NPs to introduce exogenous genes into MSCs due to the lower risk of NPs compared with viral vectors. Secondly, as drug carriers, NPs could effectively protect MSCs from direct interaction with chemotherapy drugs and control the drug release profile of NPs–MSCs. Thirdly, some NPs exhibit a distinct promoting effect on the proliferation and migration of MSCs, which are beneficial to successful cell-based drug delivery. In the past few years, progress has been made in the combination of MSCs and NPs for treating cancer though the combined system is still in its infancy stage. Therefore, an overview of the current paradigm of the novel combined tumor-targeted drug delivery system is urgently demanded to provide valuable information for future clinical transformations of this combined drug delivery system.
1. Introduction
Cancer is a major public health problem in the United States and many other parts of the world. It is currently the second leading cause of death in the United States, and is expected to surpass heart disease as the leading cause of death in the next few years.1 Even though many traditional methods including chemo-radiotherapy and surgical resection have been applied in cancer therapy, the average survival rate is very low. Patients mainly die of treatment-associated complications due to the lack of cancer-specific delivery of drugs.Several efforts have been made to develop cancer-targeted drug delivery systems.2 Among them, the emergence of NPs drug delivery systems has offered a new chapter in obtaining efficient and safe drug delivery. Owing to the special tumor microenvironment which is characterized by leaky vessel walls and the lack of lymphatic function, NPs are able to passively target tumor tissue, which is commonly referred to as enhanced permeability and retention (EPR) effect.3 Additionally, modification of NPs with various moieties is helpful in controlling pharmacokinetics of NPs, thus enhancing the tumor-targeted efficiency through active targeting effect.4 Moreover, drugs can be released from NPs through leaching, permeating and diffusing from matrix of NPs in response to external stimulus.5 In other words, drug-loaded on NPs usually present controlled-release profile which further ensure the safety of normal tissue. Above features render NPs superiorities in cancer-targeted drug delivery including: (1) improving the stability of drugs; (2) protecting nucleotides from degradation and facilitating their entry into nucleus; (3) prolonging the duration of drug action; (4) allowing less dosage of administration and relieving or avoiding side effect. Herein, NPs have promising applications in cancer-targeted delivery for various drugs, such as peptide/protein,6 DNA/RNA7 and chemodrugs.8
However, applications of NPs are not so extensive because of the deficiency of their clinical performance. Firstly, clearance of NPs from blood stream by reticuloendothelial system (RES) leads to premature release of drug cargo.9 Secondly, dense network of collagen fibers and high interstitial fluid pressure hinder the homogeneous distribution of NPs and cause NPs, almost accumulating at perivascular region of tumor.10 Therefore, although NPs have been reported to deliver more drugs to tumor tissue than the same drugs in a solution formulation, NP-based drug delivery may be better called “improved delivery” since only 10% of the injected dose arrives at the target tissue.11
MSCs possess many attractive inherent properties and would present as a solution to overcome the above limitations. They could circumvent the immunosurveillance by modulating immune reaction.12 Also, they have the ability to migrate within tumor tissue, providing increased penetration of NPs into the interior of tumor.13 Notably, NP-loaded cell carriers could promote NPs retention in tumor tissues.14 On the other hand, NPs drug delivery system may also benefit MSC-mediated drug delivery system in many aspects. For instance, NPs play irreplaceable role in gene-engineering of MSCs due to their unique properties, such as high loading capacity, high safety and easy manipulation. Moreover, “smart” NPs are designed to release drug cargo under tumor-specific trigger or external stimuli, such as heat,15 low pH, enzyme and light.16 This controlled release profile could ensure vitality of drug-loaded MSCs before they have arrived at tumor tissue. Until now, NPs such as silica,17 gold,18 silver,19 quantum dots,20 iron oxide21 and cationic polymer based NPs22 have been widely studied in MSCs tracking or engineering. These investigations pave the way for the combination of MSCs with drug-loaded NPs for anticancer drug delivery. This article summarizes main properties of MSCs related with tumor-targeted drug delivery and promising applications of the combination of MSCs and drug-loaded NPs in gene and chemotherapy (Table 1). We also review the interaction between MSCs and NPs, such as the ways to improve cellular uptake of NPs and to keep the vitality of MSCs after internalization of anticancer drug-loaded NPs. In the end, potential challenges and future directions of combined drug delivery system are discussed.
| Properties of MSCs | Benefits in MSC-based cancer therapy |
|---|---|
| Downregulation of survival signaling23 | Synergistic effect of inhibiting tumor cell proliferation with drugs |
| Easy isolation and expansion in vitro | Available for large scale production |
| Hypoimmunogenicity24 | Suitable for multiple doses and allogeneic MSCs |
| Tumor-tropism | Tumor-targeted delivery of anticancer drugs |
| Expression of known receptors on cell surface25 | Modification of NPs with related ligand to improve their affinity to MSCs |
| Nonsensitive to cytotoxic agent26 | Acting as chemodrug carriers without suffering serious toxic effect |
2. Mesenchymal stem cells
MSCs exist in the numerous adult tissue stroma and could differentiate to various tissue cells via specific induction. Although MSCs have no specific surface markers to distinguish themselves from other stromal cells, high expression of certain surface markers such as CD73, CD90, CD105 and no expression of hematopoietic lineage surface markers have been set as a standard of MSCs identification.27 MSCs are anchorage-dependent cells, thereby making their isolation fairly straightforward. Remarkably, there is no expression of major histocompatibility complex II and only a small amount expression of major histocompatibility complex I on MSCs membrane. Besides, they possess unique immuno-modulatory properties such as suppressing cytokine secretion of T-cells and proliferation of NK cells.28 Therefore, no host immunoresponse is evoked upon allotransplantation. The properties of MSCs, such as pluripotency, resource availability, immune privilege as well as free of ethical issue have gifted MSCs promising application in cell transplantation and tissue engineering. For instance, MSCs have been investigated as a novel strategy for T cell-mediated diseases such as graft-versus-host disease,29 Crohn's disease,30 diabetes,31 organ transplantation rejection32 and tissue repair of lung,33 heart,34 kidney35 and liver.36 During the therapy of tissue injury, MSCs displayed tropism to lesion location by intravenous administration.37 Meanwhile, cancer is often referred as “wound never healed”.38 Likewise, the following studies demonstrated that MSCs have the ability not only to selectively home and engraft to solid tumors, such as gliomas,39 lung and breast cancer,40 ovarian cancer,41 melanomas,42 hepatoma43 and prostate cancer,44 but also to actively scout out metastases far away from the primary site of the tumor.45 This discovery takes MSCs into the field of tumor-targeted delivery of therapeutic and diagnostic reagents.2.1. Mechanisms of tumor-tropism of MSCs
That MSCs could act as “magic bullets” to various tumor region has been proved. However, the mechanisms behind their tumor tropism are difficult to be clarified. Traditionally, homing progress may involve two steps (Fig. 1): firstly, shear resistant adhesion between circulating cells and the vascular endothelium at the target tissue, which is mediated by “homing receptors” expressed on cells and relevant endothelial co-receptors, leads to cell-tethering and rolling contacts on the endothelial cell surface46 (Step 1); secondly, chemokines trigger the activation of integrin adhesiveness, followed by firm adhesion and extravasation47 (Step 2). | ||
| Fig. 1 The progress of MSCs homing to tumor area. | ||
Tumors have been compared to unhealed wounds that produce continuous source of inflammatory factors which are potentially responsible for recruitment and engraftment of MSCs in tumor site. Extensive studies have shown that migration of MSCs is influenced by a large range of receptor pairs including SDF-1/CXCR4, HGF/c-Met, VEGF/VEGFR, PDGF/PDGFR, MCP-1/CCR2, and HMGB1/RAGE.48 Among these receptor pairs, CXCR4 receptor and its chemokine, SDF1/CXCR4, plays the most important role in the regulation of MSCs migration, along with mediating tumor invasion and metastasis.49 Neutralization of CXCR4 on MSCs using anti-CXCR4 monoclonal antibody would lead to significant decrease in homing ability of MSCs.50 Up to now, many of inflammatory mediators secreted by tumor cells have been proved to be involved in recruitment of MSCs to tumor sites. However, tumor-tropism is dependent on a multitude of signals which still remain to be uncovered.
2.2. Strategies to enhance tumor-tropism of mesenchymal stem cells
Treatment efficacy of MSC-mediated cancer therapy is related to the level of MSC accumulation at tumor sites. Unfortunately, MSCs may lose the expression of CXCR4 in culture medium, resulting in much lower tropism to tumor tissue.51 In order to deliver a therapeutical effective dose of drug to tumor site, methods have been developed to enhance tumor tropism of MSCs. Firstly, radiation increase the expression of inflammatory factors which mediate the recruitment of MSCs into tumor sites. To confirm that, Klopp et al. unilaterally irradiated bilateral murine 4T1 breast carcinomas for 48 h. The level of MSCs engraftment in limb receiving radiation were 34% higher than that of contralateral unirradiated limb, which was relevant to up-regulation of CC motif chemokine receptor-2 (CCR2) of MSCs.52 Secondly, pre-activation MSCs with tumor-necrosis factor-α (TNF-α) or interleukin-1β (IL-1β) will significantly improve homing of MSCs, and this increased adhesion could be completely blocked by pretreating with anti-vascular cell adhesion molecule-1 (anti-VCAM-1) monoclonal antibodies.53 Thirdly, MSCs transfected with epidermal growth factor receptor (EGFR) showed increased migration towards mouse glioma compared with untreated MSCs.54 In other words, enhancement of MSCs tropism could be achieved through varying the expression of homing receptor by gene modification and pre-activation. Fourthly, cell source is a determine factor in cell homing ability on account of their intrinsic characteristics. For example, MSCs are more suitable as drug delivery vehicle for organs, such as breast and ovaries, while neutral stem cells (NSCs) might be the preferred cell carriers for central nervous system malignancies.55 Fifthly, the level of MSC accumulation in tumor site is a function of synergetic action of tumor environment and administration route.56 MSCs have been used to treat various types of tumor through different administration routes including intravenous,57 intra-arterial,39 intraperitoneal injection58 and intracerebral injection.59 Compared with intravenous injected cells, intra-tumor administrated cells have lower intratumoural accumulation as they are more likely to die when located within a hypoxic tumor microenvironment.57 MSCs delivered via nasal route could arrived at glioma lesion quickly as a result of fast penetration from nasal cavity into brain tissue.60 In fact, intravenous administration may be favorable for the treatment of lung tumors since most of intravenously injected MSCs are trapped within the lungs capillaries.61 Additionally, magnetic targeting using NPs to elevate tumor tropism of cell carriers may provide an alternative to biological methods. For instance, FDA-approved ferumoxytol NPs facilitate engraftment of magnetically-targeted stem cell into the focus area and lead to augmented therapeutic benefit under magnetic field simulation.62 Tumor-tropic efficiency is critically important for optimizing and designing MSC-based drug delivery platform. The above ways to improve cell migration are based on the mechanism of cell homing. Hence, fully understanding of the complicated pathways involved in MSCs homing and engraftment to tumor area will benefit therapeutic efficiency of this novel drug delivery platform.633. Interaction of NPs and MSCs drug delivery systems
MSCs have been widely studied as a cancer-targeted drug delivery vehicle due to hypoimmunogenic and active targeting properties. However, the side effect of chemodrugs and viral gene vectors on cell carriers is a major limitation of MSC-based drug delivery. Recently, a novel drug delivery strategy has been investigated to guarantee viability and function of MSCs via combination of drug-loaded NPs. However, the mechanisms of internalization of NPs, the distribution of NPs in MSCs and the influence of NPs on the fate of MSCs should be well studied prior to clinical trials. In the following sections, we will survey the interaction between MSCs and NPs, as well as their synergistic effect and potential challenges in anti-tumor therapy.3.1. Cellular uptake of NPs by MSCs
Incorporating enough drug-loaded NPs into MSCs is necessary to achieve sufficient drug concentration in tumor tissue. Various mechanisms involve in internalization of NPs into MSCs including nonspecific endocytosis and receptor-mediated endocytosis.64 The extent of endocytosis is dramatically affected by physicochemical properties of NPs.65 For a given kind of NPs, the rate and dose of cellular uptake vary with the size of NPs, the larger ones getting into the cells much more slowly than the smaller ones.66 Besides, surface charge is another decisive factor in the internalization of NPs. Positively charged materials which can insert into and cross biologic cell membrane through electric attraction have been widely used to facilitate cellular uptake of NPs.67 Internalization of NPs into MSCs also could be facilitated via receptor-mediated uptake. Ligand modified NPs show increased probability of adhesion with cell membrane by binding with related receptor on the cell surface. Apart from the characteristics of NPs, the efficiency of cellular uptake is positively associated with the rate of cell proliferation. The highest uptake efficiency is obtained between the second and fourth passages when cells are growing and expanding rapidly.68 Moreover, cellular uptake efficiency may be improved by proper incubation with MSCs. As we all know, NPs are internalized in a time dependent manner and successful cell uptake assay should be conducted in serum-free medium.69 However, MSCs is sensitive to culture medium. Serum deprivation has an adverse effect on cell vitality in time-dependent manner. Hence, the incubation time should be well studied to balance the degree of cellular uptake and cell viability.3.2. Location and distribution of NPs in MSCs
After internalization, NPs are usually located inside of lysosome and destroyed there. Nevertheless, chemotherapeutics released from NPs would impair cell vitality, especially their tumor migratory property. Therefore, keeping drug-loaded NPs from lysosomal degradation before MSCs arriving at targeted area is important in achieving successful cancer-targeted drug delivery. As for gene-loaded NPs, endosomal escape is a necessity for protecting therapeutic genes from enzymatic degradation. Hence, NPs designed for the combined system should resist to lysosomal degradation. Materials possessing lysosome escape ability can be classified into two groups according to different escaping mechanisms: raising the osmotic pressure of lysosome and fusing with lysosomal membrane. For example, polymers with abundant amino such as polyethyleneimine (PEI), PAMAM have the ability to absorb large amounts of hydrogen ions under acidic condition, thereby resulting in rapid increase in osmotic pressure within the lysosomes and leading to lysosomal rupture. This phenomenon is also called “proton sponge” effect. Lipids having membrane fusion ability, e.g. 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), are widely applied to facilitate endosome escape of NPs.70 Additionally, some polypeptides, including influenza virus, influenza virus hemagglutinin-2 and hornets peptide, change conformation to the helical structure under acidic condition, which is easily fused with membrane and decrease the stability of lysosomal membrane structure.71 Besides, photochemical internalization (PCI) is a novel technology based on the photosensitizers located in endocytic vesicles that induces the release of endocytosed NPs into cytosol upon activation by light (Fig. 2).72 It has been applied to facilitate the cell uptake and transfection efficiency of NPs for MSCs.73 Another strategy to avoid lysosome degradation is direct cytosolic delivery. Recombination high density lipoprotein (rHDL) could bind with scavenger receptor class B type I (SR-BI) which mediates the direct cytosol lipid transport, presumably by forming a hydrophobic channel in the cell membrane.74 Up to now, rHDL has been frequently investigated as a vector for cancer-targeted delivery of DNA and siRNA. These reports lay solid foundation for future application of rHDL in MSCs engineering.75 | ||
| Fig. 2 Schematic concept of photosensitizer-induced uptake and endosomal escape. | ||
After entering into cytoplasm, gene vectors may firstly scatter in the cytoplasm and should finally enter the nucleus for transfection.76 However, it is well established that nuclear pores do not allow the spontaneous diffusion of macromolecules from cytoplasm into cell nucleus. In order to improve the efficiency of nuclear transport, nuclear localization signal, such as transcription factors, histone, glycoproteins and hormone, are investigated as nuclear-target ligand for gene vector. It has been shown that glucocorticoid-modified NPs display enhanced efficiency of gene delivery by expanding nuclear pores.77
Although mechanisms involved in the anticancer drugs unloading from MSCs carriers at the tumor site remain to be investigated, we can learn from previous reports about the ways in which drug release from other types of cell carriers. It is possible that drugs may be discharged from the cell carriers through both passive diffusion, active transport or released from apoptotic MSCs which is induced by cytotoxic cargo78 (Fig. 3). Moreover, internal and external stimulation such as increasing concentration of intracellular Ca2+ and mild hypothermia, have been applied to modulate the dose and site of drug unloading from mononuclear phagocytes.79 Recently, in order to facilitate efflux of drugs, MSCs are deliberately transfected with the multidrug resistance gene which encodes ATP-binding cassette transporters. This manipulation increase chemotherapeutic resistance of MSCs, which is also favorable for maintaining the cell viability.80
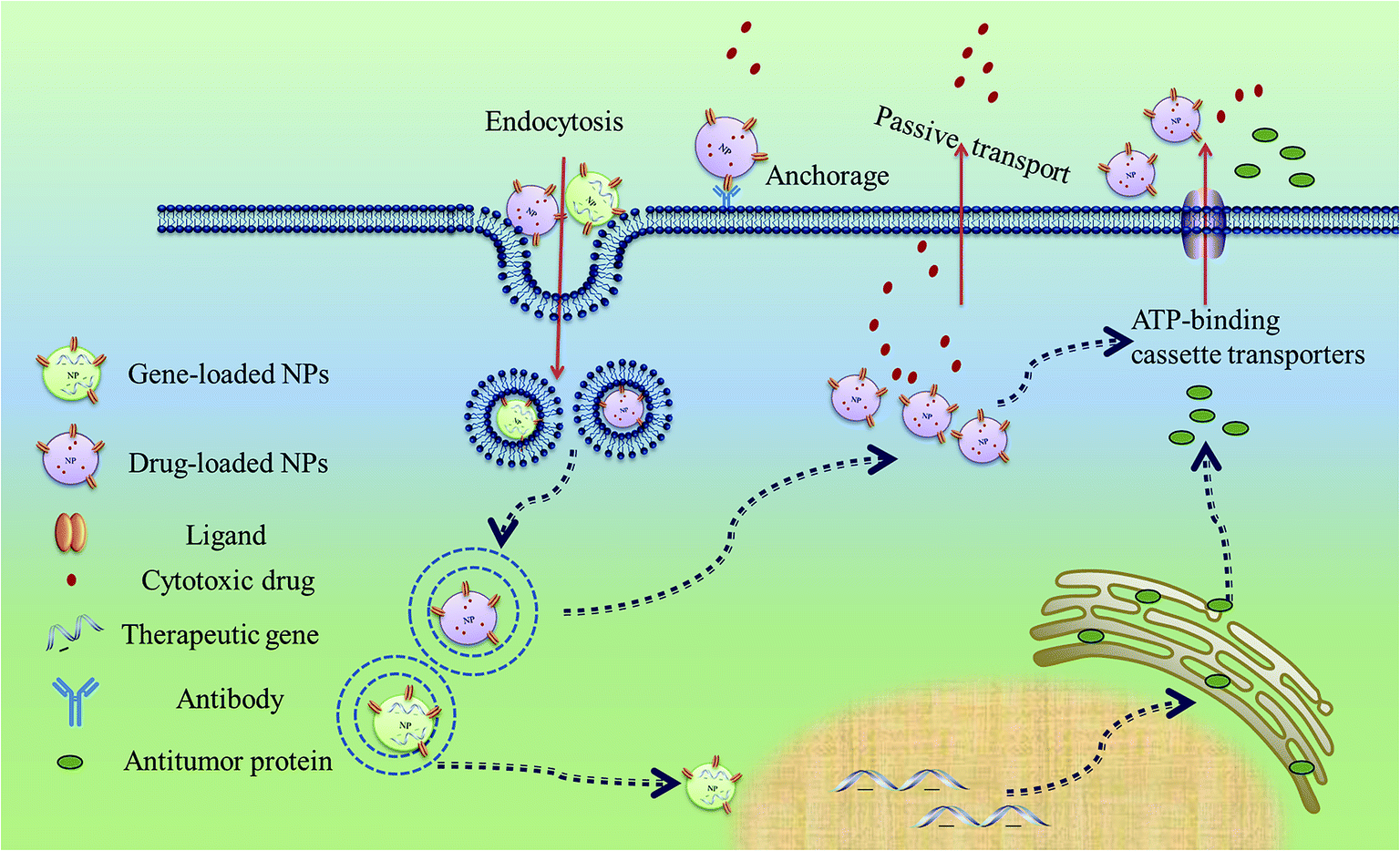 | ||
| Fig. 3 Interaction between MSCs and NPs. | ||
3.3. Effects on proliferation and migration of MSCs
Migration ability and proliferation rate of MSCs are important indexes of successful cell-mediated drug delivery. It is necessary to evaluate the influence of NPs on MSCs to confirm the feasibility of the combined drug delivery system. Standard MTT assay was used to test cytotoxicity of hyaluronic acid-based polymer mesoporous silica (HA-MSNs) on MSCs. Fire fly luciferase expressing MSCs (fluc-MSCs) model was established to study the influence of NPs on protein expression. Results demonstrated that internalization of HA-MSNs into MSCs did not affect viability and intracellular protein expression.81 Additionally, ferucarbotran, an ionic superparamagnetic iron oxide particle, was shown to promote cell proliferation by diminishing intracellular H2O2.82 Similarly, iron-based magnetic NPs could actively increase chemokine receptor CXCR4 expression of MSCs, thereby improving cell homing efficiency.83 On the contrary, the presence of AuNPs in MSCs leads to increased population but decreased cell motility. The degree of the influence on MSCs depended on sizes of AuNPs, where smaller particles required a sevenfold higher concentration to achieve the same effect as larger ones.84 Many researches suggested that the impacts of NPs on MSCs, such as proliferation, protein expression and migration, which may vary according to the physicochemical property.85 Hence, in order to reduce toxicity of NPs and improve biological activity of MSCs, the type, concentration and modification methods of NPs should be selected with discreet.4. Combination of NPs and MSCs in cancer-targeted drug delivery
4.1. Combination of NPs and MSCs in cancer-targeted gene therapy
Gene therapy is a great breakthrough in anticancer medicine for its unlimited range of therapeutic efficiency.86 However, gene therapy fails to achieve its full potential due to quick elimination of therapeutic DNA by immune system. This obstacle may be resolved by gene engineered MSCs which are gifted with tumors tropism ability and immune privilege. MSC-mediated anticancer therapy mainly involves eight types of genes: interleukins, interferons, prodrug activators, oncolytic viruses, pro-apoptotic proteins, antiangiogenic agents, growth factor antagonists and antibodies which have been reviewed in another article.48 Successful gene transfection of MSCs relies on gene vectors with low toxicity and high transfection efficiency. Although viral vectors may present high transfection ability and allow for stable gene expression, their clinical applications are limited as result of some potential problems, such as oncogenic transformation, pathogenic risk, and induction of immune responses.87 The risks associated with their use lead to increasing investigations of non-viral vectors to attain the same purpose, even if MSCs are difficult to be transfected in this way (Table 2).| MSCs type | NPs type | Cargo | Size | Zeta potential | Findings | Reference |
|---|---|---|---|---|---|---|
| hMSCs | PLGA | PTX | 259 nm | −17.1 mV | Therapy of lung tumor | 69 |
| BMSCs | CTS-PLGA NPs | PTX | 142 nm | 9.22 mV | Deep analysis of the interaction between PTX-loaded NPs and MSCs | 78 |
| BMSCs | Iron oxide MNPs | None | None | None | Improvement of tumor tropism and noninvasive imaging of MSCs | 83 |
| BMSCs | PEI600-β-CyD | TRAIL | Around 200 nm | Around 40 mV | Inhibiting proliferation of lung metastases | 88 |
| TK-BMSCs | SP | Ganciclovir | Around 100 nm | 40 mV | Pulmonary metastasis therapy | 89 |
| BMSCs | SP | IL-12 | Around 230 nm | 15 mV | Reducing lung metastasis | 90 |
| BMSCs | SP and prodrug-loaded liposomes | Suicide gene | Around 938 nm | None | Selectively eradicating lung metastasis and minimizing systemic toxicity | 91 |
| hMSCs | Neutravidin-coated NPs | None | 40 nm | None | As cellular patches for stem cell-based cancer therapy | 92 |
| BMSCs | Silica nanorattle | Dox | 125.6 nm | 35 mV | Increased and prolonged intratumoral retainment of drug | 93 |
| BMSCs | Fluorescent magnetic NPs | None | 50 nm | 32.25 mV | Hyperthermia therapy and targeted imaging of early gastric cancer | 94 |
| BMSCs | Polymethyl methacrylate | Porphyrin | 39 nm | 28.5 mV | Therapy of osteosarcoma | 95 |
| MIAMI cells | Poly-lactic acid (PLA) | Coumarin-6 | 136 nm | −2.11 mV | Simple proof of the combination of NPs and MSCs | 96 |
| Lipid nanocapsules (LNCs) | 88 nm | −3.7 mV | ||||
| Lipid nanocapsules | Fc-diOH | Around 76 nm | −7 mV | Therapy of brain tumor | 97 |
To overcome the efficiency–cytotoxicity dilemma, several methods have been tried to improve the physicochemical and biological properties of PEI. Chemical modification of PEI with hydrophobic ligand has been proved as an effective way to reduce cytotoxicity of PEI by decreasing the density of positive charge of PEI.101 Moreover, covalent combination of PEI and hyaluronic acid (HA, a natural ligand for receptors on MSCs) also actively improved biocompatibility and internalization kinetics of PEI. Moreover, conjugating negatively charged HA to PEI resulted in improved release of DNA because of the loose association between DNA with PEI–HA, which is conducive to successful gene delivery.102 Three-dimensional (3D) cell cultures mimic the situation in vivo by embedding cells in a defined scaffold which simulates the extracellular matrix of structural proteins and other biological molecules. This biomaterial scaffolds may alleviate cytotoxicity of PEI by boosting cell proliferation. Besides, the increased cellular attachment, proliferation and motility throughout the 3D collagen allowed MSCs to be exposed to more PEI/DNA complexes and finally resulted in sustained and elevated levels of gene expression.103 Taken together, these studies provided insight in enhancing transfection efficiency and reducing cytotoxic effect of PEI, which would greatly accelerate its application in MSC-engineering for future clinical studies.
Serum deprivation is a prerequisite for optimal PEI-mediated transfection since negatively charged serum proteins destabilize of the PEI/DNA polyplexes. However, MSCs may loss vitality in serum-free culture medium, which would go against successful therapeutic applications. To solve this problem, lipopolyplexes were developed by coating PEI/DNA polyplexes with serum-resistant cationic lipid sterols for MSC transfection. Serum-resistant cationic lipid sterols improved the performance of PEI/DNA complexes as gene vectors in the presence of serum.107
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 Da, which is three times higher than that of lipofectamineTM2000.113 The high ability of gene delivery coupling with low toxicity clearly indicates that SP is a promising gene vector for MSCs. Traditionally, serum deprivation is a prerequisite for cellular uptake of NPs to avoid the neutralization of cationic polymer by serum. However, the safety requirements for clinical application as well as the notable sensitivity of MSCs to in vitro culture conditions demand the development of transfection performing in growth medium (i.e., serum- and growth factor-containing). Recently, a unique type of negatively charged gene vector, termed as anioplex, was prepared by mixing plasmid DNA with SP, at a SP nitrogen: DNA phosphate (N:P) ratio of 1:1. Anioplex allows effective transfection in the presence of serum and its transfection efficiency for MSCs was comparable to or exceeded Lipofectamine 2000.114
300 Da, which is three times higher than that of lipofectamineTM2000.113 The high ability of gene delivery coupling with low toxicity clearly indicates that SP is a promising gene vector for MSCs. Traditionally, serum deprivation is a prerequisite for cellular uptake of NPs to avoid the neutralization of cationic polymer by serum. However, the safety requirements for clinical application as well as the notable sensitivity of MSCs to in vitro culture conditions demand the development of transfection performing in growth medium (i.e., serum- and growth factor-containing). Recently, a unique type of negatively charged gene vector, termed as anioplex, was prepared by mixing plasmid DNA with SP, at a SP nitrogen: DNA phosphate (N:P) ratio of 1:1. Anioplex allows effective transfection in the presence of serum and its transfection efficiency for MSCs was comparable to or exceeded Lipofectamine 2000.114Chitosan (poly-(β-1/4)-2-amino-2-deoxy-D-glucopyranose, CS) is the N-deacetylated derivative of chitin with high biocompatibility in spite of the presence of high percentage of nitrogen which can effectively bind with DNA through electrostatic attraction. The transfection potential is firstly evaluated on MSCs, human osteosarcoma cells (MG63) and human embryonic kidney cells (HEK293). Transfection efficiency of CS is much lower than LipofectamineTM2000 regardless of the cell type, the molecule weigh of CS and the mount of DNA.115 The low ability of gene transfection is due to the instability of CS/DNA complexes and low pH buffering ability.116 Incorporation of PEI into CS-NPs would provide high concentration of protonable amine on the NPs and enhance dispersing stability of CS-NPs. Besides, CS/PEI-NPs showed higher transfection efficiency than PEI itself due to the lower toxicity of CS/PEI-NPs as indicated by the high percentage of cell viability.117
4.2. Combination of NPs and MSCs in cancer-targeted chemodrug therapy
MSCs could increase chemosensitivity of breast cancer cells to adriamycin and 5-fluorouracil. Hence, it is desirable to use MSCs as a vehicle for chemodrug in cancer therapy.120 However, side effects of chemodrug on MSCs hamper this therapeutic application. Several strategies using NPs have been tried to overcome this limitation (Table 2). Roger et al. tentatively incorporated coumarin-loaded polylactic acid NPs (PLA-NPs) and lipid nanocapsule (LNCs) into MSCs. Results demonstrated that these two types of NPs could be uptaken by MSCs and had no negative effect on biological function of cell carriers, theoretically confirming the possibility of the combination of MSCs and NPs for chemodrug delivery.96 The follow-up study showed that the internalization of 2-ferrocenyl-1,1-bis(4-hydroxyphenyl)-but-1-ene (Fc-diOH) loaded LNC did not induce apoptosis of MSCs irrespective of the drug concentration, while viability of cancer cells decreased to less than 65% at the concentration of 100 μg mL−1 Fc-diOH-LNCs. The results further confirmed that Fc-diOH-loaded LNCs is a safe and effective cargo for MSC carriers.97 Photodynamic therapy (PDT) is a clinically approved method using photosensitizer for tumor therapy, which can lead to serious cell necrosis only when exposed to sufficient light irradiation.121 Therefore, PDT has the potential to circumvent main barrier in MSCs-based chemodrug delivery. Moreover, NPs pre-loaded with a photosensitizer, namely meso-tetrakis(4-sulfonatophenyl)porphyrin (TPPS), could be uploaded into MSC with high compatibility and trigger osteosarcoma (OS) cell death in vitro upon specific photoactivation.95 Many chemodrugs, such as doxorubicin (Dox), could induce cell apoptosis only when they arrive at the nucleus. Therefore, anchoring drug-loaded NPs on the cell membrane could avoid the trafficking of chemodrugs to into the nucleus of MSCs.122 Antibody–antigen recognitions, surface thiols and MSC affinity peptide could act as a linker between surface of MSCs and NPs or chemodrugs.123 MSCs engineered by surface-conjugated Dox-loaded silica nanorattle significantly increased and prolonged intratumoral distribution Dox. Also, silica nanorattle engineered MSCs released Dox in response to relative low pH in tumor environment, which further insured the viability of cell carriers before circulating to tumor site.93 Moreover, viability of MSCs was higher than 90% even at a high Dox concentration of 16 μg mL−1. The result was consistent with another report that MSCs were not sensitive to 5-FU compared with tumor cells. It may attribute to the overexpression of ATP binding cassette (ABC) transporters that mediate drug efflux, thereby providing the fundamental basis for MSCs as chemodrug carriers.58 Investigations thus far using NPs to avoid the cytotoxicity of chemodrug on MSCs are proof-of-concept trials. More work must be done to show the superiority in therapeutic efficiency of combined drug delivery system compared with free-standing drug delivery systems.5. Conclusion and outlook
Two different strategies are utilized in alliance of MSCs and NPs for cancer-targeted drug delivery. First, MSCs are genetically engineered by NP-based gene vector to produce antitumor protein. Second, cytotoxic agent-loaded NPs are incorporated into MSCs or anchored on the cell surface. Then, NPs engineered MSCs act as “Trojan horses” to deliver the therapeutics to targeted sides (Fig. 4). | ||
| Fig. 4 Schematic illustration of combination of NPs and MSCs drug delivery systems for cancer therapy. | ||
The combination of MSCs with NPs offers us a more safe and effective drug delivery system by overcoming hurdles encountered when each one is used alone. However, this new drug delivery system still faces some challenges. MSCs are reported to play dual roles in tumorigenesis.124 The function of MSCs on tumor progression varies from cancer types and administration routes (Table 3).
| Cell source | Tumor model | Results | Reference |
|---|---|---|---|
| AD-MSCs | EOC | AD-MSCs co-intraperitoneally injected with EOC significantly promoted EOC growth and metastasis | 125 |
| hMSCs | A549 | MSCs co-subcutaneously injected with A549 or CL1-5 promoted the formation of tumor | 126 |
| BMSCs | DU145 | DU145 show increased motility when co-cultured with BMSCs | 127 |
| BMSCs | RCAS-Neu | MSCs administered by tail vein after or co-implanted with RCAS-Neu cells with different ratios have no effect on tumor progression | 128 |
| hMSCs | SGC-7901 | The growth of SGC-7901 cells could be suppressed by subcutaneous co-injections with hMSCs | 129 |
| AD-MSCs | MCF-7 | ASCs co-cultured with and suppressed the growth of MCF-7 cells | 130 |
| BMSCs | Colitis-associated tumorigenesis | MSCs injected via the tail vein ameliorated the tumorigenesis | 131 |
| BMSCs | Subcutaneous glioma models | Co-administration of MSCs with glioma cells resulted in significant reduction in tumor volume and vascular density | 132 |
The accumulation of genetic mutations during cell culture and the subsequent risk of cell transformation is another drawback of MSC-based therapy.133 Although introduction of MSCs with a backup suicide gene which carries a regulatable promoter to selectively eradicate misbehaving cells may partly resolve the problem to some extent, exhaustive investigations should also be carried out to monitor chromosomal aberrations in MSCs prior to any clinical application.134 Moreover, variations in isolation methods and cell passage numbers may also influence cell homing ability and behavior of MSCs within the tumor microenvironment, thus affecting dose and duration of therapeutics at the tumor site. The development of well-characterized immortalized clonal stem cell lines may ease the constraints in practical application of MSCs. However, enhanced potential of unwanted changes in MSCs functional phenotype such as gain of tumorigenic potential or loss of specific migration could not be ignored. Therefore, the standard method of in vitro culture of MSCs and quality criterion of drug-loaded cell carriers are necessary to be established as guidance for the industrialized production.
Regarding the drug-loaded NPs, their formulations should be optimized for the industrial utilization and clinical translation of MSC-based drug delivery system. Fundamentally, the ideal drug carriers for cell-mediated delivery should present controlled drug release profile with high biocompatibility. Additionally, it is beneficial to incorporate drugs and imaging agents within the same NPs for non-invasive real-time visualization of MSCs. Furthermore, relative low drug-loading capacity is a defect of MSC-based delivery system. In order to overcome the defect, strategies to increase the drug loading capacity and cellular uptake of NPs are needed to be developed. Meanwhile, the upper limit should be set with discreet to avoid drug cytotoxic effects on the cell-carriers. It is worth noting that drug-loaded NPs for MSC-mediated delivery system may differ from conventional cancer-targeted NPs. For example, NPs formulated for cancer-targeted gene delivery usually present uniform diameters about 100 nm to achieve maximum exploration of EPR effect. Nevertheless, particle size of NPs constructed for MSC-engineering is not so restricted as long as NPs exhibit high transfection ability for MSCs. Besides, chemodrug-loaded NPs for MSC-based therapy are designed to be released from cell carriers at tumor side, while traditional NPs are aimed at maximizing uptake and the retainment in tumor cells. In this case, discovering appropriate materials and new techniques for NPs may open a new perspective of MSC-based anticancer drug delivery.
Overall, future efforts should be directed toward a better understanding of biological properties of MSCs, the mechanisms of MSCs homing to tumor areas, the interaction between NPs and MSCs, the in vivo of biodistribution MSCs–NPs and the action of the MSCs–NPs in tumor area. It would allow us to obtain a more deep understanding into the shortcomings of MSCs–NPs mediated tumor-targeted drug delivery system and would assist us in finding optimal strategies to surpass above limitations in the future.
Acknowledgements
We gratefully appreciate financial supports from the National Natural Science Foundation of China (No. 81102398 and 81273469), the Natural Science Foundation of Jiangsu Province (No. BK2011624), the Ministry of Education Doctoral Program of Higher Specialized Research Fund project (No. 20110096120003 and 20113234120008), the Fundamental Research Funds for the Central Universities (No. JKVD2013011), the Open Project Program of State Key Laboratory of Natural Medicines, China Pharmaceutical University (No. SKLNMKF201305 and SKLNMKF201215) and the National Found for Fostering Talents of Basic Science (No. J1030830).References
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2015, 65, 5–29 CrossRef PubMed.
- K. Cho, X. Wang, S. Nie and D. M. Shin, Clin. Cancer Res., 2008, 14, 1310–1316 CrossRef CAS PubMed.
- A. S. Narang and S. Varia, Adv. Drug Delivery Rev., 2011, 63, 640–658 CrossRef CAS PubMed.
- J. D. Byrne, T. Betancourt and L. Brannon-Peppas, Adv. Drug Delivery Rev., 2008, 60, 1615–1626 CrossRef CAS PubMed.
- H. Ren, L. Zhang, J. An, T. Wang, L. Li, X. Si, L. He, X. Wu, C. Wang and Z. Su, Chem. Commun., 2014, 50, 1000–1002 RSC.
- T. Jiang, W. Sun, Q. Zhu, N. A. Burns, S. A. Khan, R. Mo and Z. Gu, Adv. Mater., 2015, 27, 1021–1028 CrossRef CAS PubMed.
- C. Wang, X. Bao, X. Ding, Y. Ding, S. Abbad, Y. Wang, M. Li, Y. Su, W. Wang and J. Zhou, Polym. Chem., 2015, 6, 780–796 RSC.
- W. Tai, R. Mo, Y. Lu, T. Jiang and Z. Gu, Biomaterials, 2014, 35, 7194–7203 CrossRef CAS PubMed.
- K. Shanthi, K. Vimala, D. Gopi and S. Kannan, RSC Adv., 2015, 5, 44998–45014 RSC.
- H. Kobayashi, R. Watanabe and P. L. Choyke, Theranostics, 2013, 4, 81–89 CrossRef PubMed.
- I. K. Kwon, S. C. Lee, B. Han and K. Park, J. Controlled Release, 2012, 164, 108–114 CrossRef CAS PubMed.
- A. Sudeepta and M. F. Pittenger, Blood, 2005, 105, 1815–1822 CrossRef PubMed.
- R. Liu, Cancer Lett., 2014, 353, 145–152 CrossRef CAS PubMed.
- R. Mooney, Y. Weng, R. Tirughana-Sambandan, V. Valenzuela, S. Aramburo, E. Garcia, Z. Li, M. Gutova, A. J. Annala and J. M. Berlin, Future Oncol., 2014, 10, 401–415 CrossRef CAS PubMed.
- K. Qian, Y. Ma, J. Wan, S. Geng, H. Li, Q. Fu, X. Peng, X. Kan, G. Zhou and W. Liu, J. Controlled Release, 2015, 41–49 CrossRef CAS PubMed.
- Y. Tian, Y. Kong, X. Li, J. Wu, A. C.-T. Ko and M. Xing, Colloids Surf., B, 2015, 147–155 CrossRef PubMed.
- X. Huang, F. Zhang, H. Wang, G. Niu, K. Y. Choi, M. Swierczewska, G. Zhang, H. Gao, Z. Wang and L. Zhu, Biomaterials, 2013, 34, 1772–1780 CrossRef CAS PubMed.
- N. Kawazoe and G. Chen, Biomaterials, 2015, 54, 226–236 CrossRef PubMed.
- C. Y. Sun, Y. J. Che and S. J. Lu, Biotechnol. Lett., 2014, 37, 467–473 CrossRef CAS PubMed.
- Y. Lei, H. Tang, L. Yao, R. Yu, M. Feng and B. Zou, Bioconjugate Chem., 2007, 19, 421–427 CrossRef PubMed.
- X.-H. Jing, L. Yang, X.-J. Duan, B. Xie, W. Chen, Z. Li and H.-B. Tan, Jt., Bone, Spine, 2008, 75, 432–438 CrossRef PubMed.
- W. Wang, W. Li, L. Ou, E. Flick, P. Mark, C. Nesselmann, C. A. Lux, H. H. Gatzen, A. Kaminski and A. Liebold, J. Cell. Mol. Med., 2011, 15, 1989–1998 CrossRef CAS PubMed.
- A. Torsvik and R. Bjerkvig, Cancer Treat. Rev., 2013, 39, 180–188 CrossRef CAS PubMed.
- K. Le Blanc, Cytotherapy, 2003, 5, 485–489 CrossRef CAS PubMed.
- A. Scherzed, S. Hackenberg, K. Froelich, K. Rak, A. Technau, A. Radeloff, C. Koehler, R. Hagen and N. Kleinsasser, Toxicol. Lett., 2013, 218, 207–214 CrossRef CAS PubMed.
- H. Cheng, C. J. Kastrup, R. Ramanathan, D. J. Siegwart, M. Ma, S. R. Bogatyrev, Q. Xu, K. A. Whitehead, R. Langer and A. Dg, ACS Nano, 2012, 4, 625–631 CrossRef PubMed.
- M. Dominici, K. Le Blanc, I. Mueller, I. Slaper-Cortenbach, F. Marini, D. Krause, R. Deans, A. Keating, D. Prockop and E. Horwitz, Cytotherapy, 2006, 8, 315–317 CrossRef CAS PubMed.
- K. L. Blanc, I. Rasmusson, B. Sundberg, C. G. Ouml, M. Hassan, M. Uzunel and O. Ringd, Lancet, 2004, 363, 1439–1441 CrossRef.
- K. Le Blanc, I. Rasmusson, B. Sundberg, C. Götherström, M. Hassan, M. Uzunel and O. Ringdén, Lancet, 2004, 363, 1439–1441 CrossRef.
- D. García-Olmo, M. García-Arranz, D. Herreros, I. Pascual, C. Peiro and J. A. Rodríguez-Montes, Dis. Colon Rectum, 2005, 48, 1416–1423 CrossRef PubMed.
- L. X. Guan, H. Guan, H. B. Li, C. A. Ren, L. Liu, J. J. Chu and L. J. Dai, Exp. Ther. Med., 2015, 9, 1623–1630 CAS.
- K. Le Blanc and O. Ringdén, Biol. Blood Marrow Transplant., 2005, 11, 321–334 CrossRef CAS PubMed.
- M. Rojas, J. Xu, C. R. Woods, A. L. Mora, W. Spears, J. Roman and K. L. Brigham, Am. J. Respir. Cell Mol. Biol., 2005, 33, 145–152 CrossRef CAS PubMed.
- M. F. Pittenger and B. J. Martin, Circ. Res., 2004, 95, 9–20 CrossRef CAS PubMed.
- M. B. Herrera, B. Bussolati, S. Bruno, V. Fonsato, G. M. Romanazzi and G. Camussi, Int. J. Mol. Med., 2004, 14, 1035–1041 Search PubMed.
- B. Parekkadan, D. Van Poll, K. Suganuma, E. A. Carter, F. Berthiaume, A. W. Tilles and M. L. Yarmush, PLoS One, 2007, 2, e941 Search PubMed.
- A. Chapel, J. M. Bertho, M. Bensidhoum, L. Fouillard, R. G. Young, J. Frick, C. Demarquay, F. Cuvelier, E. Mathieu and F. Trompier, J. Gene Med., 2003, 5, 1028–1038 CrossRef PubMed.
- N. J. Oviedo and W. S. Beane, Semin. Cell Dev. Biol., 2009, 20, 557–564 CrossRef PubMed.
- A. Nakamizo, F. Marini, T. Amano, A. Khan, M. Studeny, J. Gumin, J. Chen, S. Hentschel, G. Vecil and J. Dembinski, Cancer Res., 2005, 65, 3307–3318 CAS.
- B. Sun, K.-H. Roh, J.-R. Park, S.-R. Lee, S.-B. Park, J.-W. Jung, S.-K. Kang, Y.-S. Lee and K.-S. Kang, Cytotherapy, 2009, 11, 289–298 CrossRef CAS PubMed.
- E. K. Mader, Y. Maeyama, Y. Lin, G. W. Butler, H. M. Russell, E. Galanis, S. J. Russell, A. B. Dietz and K.-W. Peng, Clin. Cancer Res., 2009, 15, 7246–7255 CrossRef CAS PubMed.
- D. Fang, T. K. Nguyen, K. Leishear, R. Finko, A. N. Kulp, S. Hotz, P. A. Van Belle, X. Xu, D. E. Elder and M. Herlyn, Cancer Res., 2005, 65, 9328–9337 CrossRef CAS PubMed.
- L. Qiao, Z. Xu, T. Zhao, Z. Zhao, M. Shi, R. C. Zhao, L. Ye and X. Zhang, Cell Res., 2008, 18, 500–507 CrossRef CAS PubMed.
- D. Kong, S. Banerjee, A. Ahmad, Y. Li, Z. Wang, S. Sethi and F. H. Sarkar, PLoS One, 2010, 5, e12445 Search PubMed.
- C. D. Porada and G. Almeida-Porada, Adv. Drug Delivery Rev., 2010, 62, 1156–1166 CrossRef CAS PubMed.
- B. Rüster, S. Göttig, R. J. Ludwig, R. Bistrian, S. Müller, E. Seifried, J. Gille and R. Henschler, Blood, 2006, 108, 3938–3944 CrossRef PubMed.
- H. Yagi, A. Soto-Gutierrez, B. Parekkadan, Y. Kitagawa, R. G. Tompkins, N. Kobayashi and M. L. Yarmush, Cell Transplant., 2010, 19, 667 Search PubMed.
- K. Shah, Adv. Drug Delivery Rev., 2012, 64, 739–748 CrossRef CAS PubMed.
- T. J. Myers, F. Granero-Molto, L. Longobardi, T. Li, Y. Yan and A. Spagnoli, Expert Opin. Biol. Ther., 2010, 10, 1663–1679 CrossRef CAS PubMed.
- M. Shi, J. Li, L. Liao, B. Chen, B. Li, L. Chen, H. Jia and R. C. Zhao, Haematologica, 2007, 92, 897–904 CrossRef PubMed.
- I. A. Potapova, P. R. Brink, I. S. Cohen and S. V. Doronin, J. Biol. Chem., 2008, 283, 13100–13107 CrossRef CAS PubMed.
- A. H. Klopp, E. L. Spaeth, J. L. Dembinski, W. A. Woodward, A. Munshi, R. E. Meyn, J. D. Cox, M. Andreeff and F. C. Marini, Cancer Res., 2007, 67, 11687–11695 CrossRef CAS PubMed.
- V. F. Segers, I. Van Riet, L. J. Andries, K. Lemmens, M. J. Demolder, A. J. De Becker, M. M. Kockx and G. W. De Keulenaer, Am. J. Physiol.: Heart Circ. Physiol., 2006, 290, H1370–H1377 CrossRef CAS PubMed.
- H. Sato, N. Kuwashima, T. Sakaida, M. Hatano, J. E. Dusak, W. K. Fellows-Mayle, G. D. Papworth, S. C. Watkins, A. Gambotto and I. F. Pollack, Cancer Gene Ther., 2005, 12, 757–768, DOI:710.1038/sj.cgt.7700827.
- A. U. Ahmed, N. G. Alexiades and M. S. Lesniak, Curr. Opin. Mol. Ther., 2010, 12, 546 CAS.
- S. Carrancio, C. Romo, T. Ramos, N. Lopez-Holgado, S. Muntion, H. Prins, A. Martens, J. Brinon, J. San Miguel and M. Del Cañizo, Cell Transplant., 2013, 22, 1171–1183 CAS.
- E. K. Sage, K. K. Kolluri, K. McNulty, S. D. S. Lourenco, T. L. Kalber, K. L. Ordidge, D. Davies, Y. G. Lee, A. Giangreco and S. M. Janes, Thorax, 2014, 69, 638–647 CrossRef PubMed.
- L. Kucerova, V. Altanerova, M. Matuskova, S. Tyciakova and C. Altaner, Cancer Res., 2007, 67, 6304–6313 CrossRef CAS PubMed.
- D. Bexell, S. Gunnarsson, A. Tormin, A. Darabi, D. Gisselsson, L. Roybon, S. Scheding and J. Bengzon, Mol. Ther., 2009, 17, 183–190 CrossRef CAS PubMed.
- I. V. Balyasnikova, M. S. Prasol, S. D. Ferguson, Y. Han, A. U. Ahmed, M. Gutova, A. L. Tobias, D. Mustafi, E. Rincón and L. Zhang, Mol. Ther., 2013, 22, 8–140 Search PubMed.
- J. Gao, J. E. Dennis, R. F. Muzic, M. Lundberg and A. I. Caplan, Cells Tissues Organs, 2001, 169, 12–20 CrossRef CAS PubMed.
- A. C. Vandergriff, T. M. Hensley, E. T. Henry, D. Shen, S. Anthony, J. Zhang and K. Cheng, Biomaterials, 2014, 35, 8528–8539 CrossRef CAS PubMed.
- R. Dwyer, S. Potter-Beirne, K. Harrington, A. Lowery, E. Hennessy, J. Murphy, F. Barry, T. O'Brien and M. Kerin, Clin. Cancer Res., 2007, 13, 5020–5027 CrossRef CAS PubMed.
- S. K. Banerji and M. A. Hayes, Langmuir, 2007, 23, 3305–3313 CrossRef CAS PubMed.
- H. Hillaireau and P. Couvreur, Cell. Mol. Life Sci., 2009, 66, 2873–2896 CrossRef CAS PubMed.
- F. Lu, S. H. Wu, Y. Hung and C. Y. Mou, Small, 2009, 5, 1408–1413 CrossRef CAS PubMed.
- A. Verma and F. Stellacci, Small, 2010, 6, 12–21 CrossRef CAS PubMed.
- D. Horák, M. Babič, P. Jendelová, V. Herynek, M. Trchová, K. Likavčanová, M. Kapcalová, M. Hájek and E. Syková, J. Magn. Magn. Mater., 2009, 321, 1539–1547 CrossRef.
- T. Sadhukha, T. D. O'Brien and S. Prabha, J. Controlled Release, 2014, 196, 243–251 CrossRef CAS PubMed.
- L. Zhu and R. I. Mahato, Expert Opin. Drug Delivery, 2010, 7, 1209–1226 CrossRef CAS PubMed.
- X. Han, B. JH, C. DS and T. LK, Nat. Struct. Biol., 2001, 8, 715–720 CrossRef CAS PubMed.
- K. Berg, P. K. Selbo, L. Prasmickaite, T. E. Tjelle, K. Sandvig, J. Moan, G. Gaudernack, Ø. Fodstad, S. Kjølsrud and H. Anholt, Cancer Res., 1999, 59, 1180–1183 CAS.
- S.-J. Park and K. Na, Biomaterials, 2012, 33, 6485–6494 CrossRef CAS PubMed.
- Z. Zhang, W. Cao, H. Jin, J. F. Lovell, M. Yang, L. Ding, J. Chen, I. Corbin, Q. Luo and G. Zheng, Angew. Chem., Int. Ed., 2009, 48, 9171–9175 CrossRef CAS PubMed.
- Y. Ding, W. Wang, M. Feng, Y. Wang, J. Zhou, X. Ding, X. Zhou, C. Liu, R. Wang and Q. Zhang, Biomaterials, 2012, 33, 8893–8905 CrossRef CAS PubMed.
- F. Zhao, Y. Zhao, Y. Liu, X. Chang, C. Chen and Y. Zhao, Small, 2011, 7, 1322–1337 CrossRef CAS PubMed.
- M. Monsigny, C. Rondanino, E. Duverger, I. Fajac and A.-C. Roche, Biochim. Biophys. Acta, Gen. Subj., 2004, 1673, 94–103 CrossRef CAS PubMed.
- T. Dai, E. Yang, Y. Sun, L. Zhang, L. Zhang, N. Shen, S. Li, L. Liu, Y. Xie and S. Wu, Int. J. Pharm., 2013, 456, 186–194 CrossRef CAS PubMed.
- E. V. Batrakov, H. E. Gendelm and A. V. Kabanov, Expert Opin. Drug Delivery, 2011, 8, 415–433 CrossRef PubMed.
- W. U. Bing, H. L. Chen and C. B. Wang, J. Mod. Oncol., 2011, 19, 248–251 Search PubMed.
- X. Huang, F. Zhang, H. Wang, G. Niu, K. Y. Choi, M. Swierczewska, G. Zhang, H. Gao, Z. Wang, L. Zhu, H. S. Choi, S. Lee and X. Chen, Biomaterials, 2013, 34, 1772–1780 CrossRef CAS PubMed.
- D.-M. Huang, J.-K. Hsiao, Y.-C. Chen, L.-Y. Chien, M. Yao, Y.-K. Chen, B.-S. Ko, S.-C. Hsu, L.-A. Tai and H.-Y. Cheng, Biomaterials, 2009, 30, 3645–3651 CrossRef CAS PubMed.
- X. Huang, F. Zhang, Y. Wang, X. Sun, K. Y. Choi, D. Liu, J.-S. Choi, T.-H. Shin, J. Cheon and G. Niu, ACS Nano, 2014, 8, 4403–4414 CrossRef CAS PubMed.
- T. Mironava, M. Hadjiargyrou, M. Simon and M. H. Rafailovich, Nanotoxicology, 2014, 8, 189–201 CrossRef CAS PubMed.
- T.-H. Chung, J.-K. Hsiao, S.-C. Hsu, M. Yao, Y.-C. Chen, S.-W. Wang, M. Y.-P. Kuo, C.-S. Yang and D.-M. Huang, ACS Nano, 2011, 5, 9807–9816 CrossRef CAS PubMed.
- M. Morille, C. Passirani, A. Vonarbourg, A. Clavreul and J.-P. Benoit, Biomaterials, 2008, 29, 3477–3496 CrossRef CAS PubMed.
- J. L. Santos, D. Pandita, J. Rodrigues, A. P. Pego, P. L. Granja and H. Tomás, Curr. Gene Ther., 2011, 11, 46–57 CrossRef CAS PubMed.
- Y.-L. Hu, B. Huang, T.-Y. Zhang, P.-H. Miao, G.-P. Tang, Y. Tabata and J.-Q. Gao, Mol. Pharmaceutics, 2012, 9, 2698–2709 CrossRef CAS PubMed.
- T.-Y. Zhang, B. Huang, Z.-Y. Yuan, Y.-L. Hu, Y. Tabata and J.-Q. Gao, Nanomedicine, 2014, 10, 257–267 CAS.
- Y.-L. Hu, P.-H. Miao, B. Huang, T.-Y. Zhang, Z.-J. Hu, Y. Tabata and J.-Q. Gao, J. Biomed. Nanotechnol., 2014, 10, 299–308 CrossRef CAS PubMed.
- T.-Y. Zhang, B. Huang, H.-B. Wu, J.-H. Wu, L.-M. Li, Y.-X. Li, Y.-L. Hu, M. Han, Y.-Q. Shen and Y. Tabata, J. Controlled Release, 2015, 209, 260–271 CrossRef CAS PubMed.
- H. Cheng, C. J. Kastrup, R. Ramanathan, D. J. Siegwart, M. Ma, S. R. Bogatyrev, Q. Xu, K. A. Whitehead, R. Langer and D. G. Anderson, ACS Nano, 2010, 4, 625–631 CrossRef CAS PubMed.
- L. Li, Y. Guan, H. Liu, N. Hao, T. Liu, X. Meng, C. Fu, Y. Li, Q. Qu and Y. Zhang, ACS Nano, 2011, 5, 7462–7470 CrossRef CAS PubMed.
- J. Ruan, J. Ji, H. Song, Q. Qian, K. Wang, C. Wang and D. Cui, Nanoscale Res. Lett., 2012, 7, 1–12 CrossRef PubMed.
- S. Duchi, G. Sotgiu, E. Lucarelli, M. Ballestri, B. Dozza, S. Santi, A. Guerrini, P. Dambruoso, S. Giannini and D. Donati, J. Controlled Release, 2013, 168, 225–237 CrossRef CAS PubMed.
- M. Roger, A. Clavreul, M.-C. Venier-Julienne, C. Passirani, L. Sindji, P. Schiller, C. Montero-Menei and P. Menei, Biomaterials, 2010, 31, 8393–8401 CrossRef CAS PubMed.
- M. Roger, A. Clavreul, N. T. Huynh, C. Passirani, P. Schiller, A. Vessières, C. Montero-Menei and P. Menei, Int. J. Pharm., 2012, 423, 63–68 CrossRef CAS PubMed.
- U. Lungwitz, M. Breunig, T. Blunk and A. Göpferich, Eur. J. Pharm. Biopharm., 2005, 60, 247–266 CrossRef CAS PubMed.
- H. H. Ahn, J. H. Lee, K. S. Kim, J. Y. Lee, M. S. Kim, G. Khang, I. W. Lee and H. B. Lee, Biomaterials, 2008, 29, 2415–2422 CrossRef CAS PubMed.
- W. J. King, N. A. Kouris, S. Choi, B. M. Ogle and W. L. Murphy, Cell Tissue Res., 2012, 347, 689–699 CrossRef CAS PubMed.
- X.-A. Chen, L.-J. Zhang, Z.-J. He, W.-W. Wang, B. Xu, Q. Zhong, X.-T. Shuai, L.-Q. Yang and Y.-B. Deng, Int. J. Nanomed., 2011, 6, 843–853 CAS.
- A. Saraf, M. C. Hacker, B. Sitharaman, K. J. Grande-Allen, M. A. Barry and A. G. Mikos, Biomacromolecules, 2008, 9, 818–827 CrossRef CAS PubMed.
- E. G. Tierney, G. P. Duffy, A. J. Hibbitts, S.-A. Cryan and F. J. O'Brien, J. Controlled Release, 2012, 158, 304–311 CrossRef CAS PubMed.
- C. Lonez, M. Vandenbranden and J.-M. Ruysschaert, Prog. Lipid Res., 2008, 47, 340–347 CrossRef CAS PubMed.
- C. T. de Ilarduya, Y. Sun and N. Düzgüneş, Eur. J. Pharm. Sci., 2010, 40, 159–170 CrossRef PubMed.
- B. Bakhshandeh, M. Soleimani, M. Hafizi and N. Ghaemi, Cytotechnology, 2012, 64, 523–540 CrossRef CAS PubMed.
- H. Song, G. Wang, B. He, L. Li, C. Li, Y. Lai, X. Xu and Z. Gu, Int. J. Nanomed., 2012, 7, 4637 CAS.
- J. L. Santos, E. Oramas, A. P. Pêgo, P. L. Granja and H. Tomás, J. Controlled Release, 2009, 134, 141–148 CrossRef CAS PubMed.
- J. L. Santos, H. Oliveira, D. Pandita, J. Rodrigues, A. P. Pêgo, P. L. Granja and H. Tomás, J. Controlled Release, 2010, 144, 55–64 CrossRef CAS PubMed.
- J. L. Santos, D. Pandita, J. Rodrigues, A. P. Pêgo, P. L. Granja, G. Balian and H. Tomás, Mol. Pharmaceutics, 2010, 7, 763–774 CrossRef CAS PubMed.
- D. Pandita, J. L. Santos, J. Rodrigues, A. P. Pêgo, P. L. Granja and H. Tomás, Biomacromolecules, 2011, 12, 472–481 CrossRef CAS PubMed.
- B. A. Clements, J. Bai, C. Kucharski, L.-L. Farrell, A. Lavasanifar, B. Ritchie, A. Ghahary and H. Uludag, Biomacromolecules, 2006, 7, 1481–1488 CrossRef CAS PubMed.
- J.-I. Jo, A. Okazaki, K. Nagane, M. Yamamoto and Y. Tabata, J. Biomater. Sci., Polym. Ed., 2010, 21, 185–204 CrossRef CAS PubMed.
- D. K. Thakor, Y. D. Teng, H. Obata, K. Nagane, S. Saito and Y. Tabata, Tissue Eng., Part C, 2010, 17, 131–144 CrossRef PubMed.
- K. Corsi, F. Chellat, L. H. Yahia and J. C. Fernandes, Biomaterials, 2003, 24, 1255–1264 CrossRef CAS PubMed.
- E. Malakooty Poor, M. Baghaban Eslaminejad, N. Gheibi, F. Bagheri and F. Atyabi, Artif. Cells, Nanomed., Biotechnol., 2014, 42, 376–384 CrossRef CAS PubMed.
- N. Pimpha, P. Sunintaboon, S. Inphonlek and Y. Tabata, J. Biomater. Sci., Polym. Ed., 2010, 21, 205–223 CrossRef CAS PubMed.
- T. Y. Zhang, B. Huang, H. B. Wu, J. H. Wu, L. M. Li, Y. X. Li, Y. L. Hu, M. Han, Y. Q. Shen and Y. Tabata, J. Controlled Release, 2015, 260–271 CrossRef CAS PubMed.
- H. Tong, C. Wang, Y. Huang, Q. Shi, J. C. Fernandes, K. Dai, G. Tang and X. Zhang, Int. J. Nanomed., 2013, 8, 1935 CrossRef PubMed.
- L. Kucerova, S. Skolekova, M. Matuskova, M. Bohac and Z. Kozovska, BMC Cancer, 2013, 13, 535 CrossRef PubMed.
- A. P. Castano, P. Mroz and M. R. Hamblin, Nat. Rev. Cancer, 2006, 6, 535–545 CrossRef CAS PubMed.
- Z. Shao, Z. Xin, Y. Pi, X. Wang, Z. Jia, J. Zhu, L. Dai, W. Chen, Y. Ling and H. Chen, Biomaterials, 2012, 33, 3375–3387 CrossRef CAS PubMed.
- M. T. Stephan, J. J. Moon, S. H. Um, A. Bershteyn and D. J. Irvine, Nat. Med., 2010, 16, 1035–1041 CrossRef CAS PubMed.
- D. Bexell, S. Scheding and J. Bengzon, Mol. Ther., 2010, 18, 1067–1075 CrossRef CAS PubMed.
- C. Chao, W. Yan, H. Michael, T. K. Hoffmann, G. Manfred, A. M. Kaufmann and A. E. Albers, PLoS One, 2011, 6, 574 CrossRef.
- H. S. Hsu, J. H. Lin, T. W. Hsu, K. Su, C. W. Wang, K. Y. Yang, S. H. Chiou and S. C. Hung, Lung Cancer, 2012, 75, 167–177 CrossRef PubMed.
- M. Barbara, L. M. Giuseppe, P. Maria Giulia, P. Pasquale and P. Claudia, J. Cell. Mol. Med., 2013, 17, 287–292 CrossRef PubMed.
- L. Usha, G. Rao, I. I. Kent Christopherson and X. Xu, PLoS One, 2013, 8, 1254–1256 Search PubMed.
- Y. Li, Y. Zhao, Z. Cheng, J. Zhan, X. Sun, H. Qian, W. Zhu and W. Xu, Exp. Mol. Pathol., 2013, 94, 430–437 CrossRef CAS PubMed.
- R. Hoon, O. Ji-Eun, R. Ki-Jong, B. Soon Koo, K. Jiye, K. Seong Joon, S. Joon Hyung, C. Eunhee, S. Ha Cheol and K. Y. Man, Cancer Lett., 2014, 352, 220–227 CrossRef PubMed.
- C. Zexian, H. Xiaowen, H. Xiaosheng, C. Xiuting, L. Xutao, Z. Yifeng, W. Xiaojian and L. Ping, Biochem. Biophys. Res. Commun., 2014, 450, 1402–1408 CrossRef PubMed.
- I. A. W. Ho, H. C. Toh, W. H. Ng, Y. L. Teo, C. M. Guo, K. M. Hui and P. Y. P. Lam, Stem Cells, 2013, 31, 146–155 CrossRef CAS PubMed.
- J.-A. Kim, K. O. Im, S. N. Park, J. S. Kwon, S. Y. Kim, K. Oh, D.-S. Lee, M. K. Kim, S. W. Kim and M. Jang, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2015, 777, 60–68 CrossRef CAS PubMed.
- F. Cao, M. Drukker, S. Lin, A. Y. Sheikh, X. Xie, Z. Li, A. J. Connolly, I. L. Weissman and J. C. Wu, Cloning Stem Cells, 2007, 9, 107–117 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |