
Photophysical, G-quadruplex DNA binding and cytotoxic properties of terpyridine complexes with a naphthalimide ligand†
Abstract
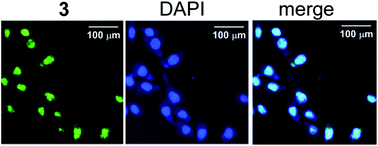
Two novel metal complexes 2–3 (metal = PdII, PtII) have been synthesized by reacting the corresponding tolylterpyridine complexes and the 4-aminonaphthalimide derivative 1. The interactions of the complexes with duplex DNA and telomeric G-quadruplex DNA have been investigated by UV-Vis spectroscopy and fluorescence spectroscopy. The studies reveal that the complexes 2–3 possess high affinity and reasonable selectivity for telomeric G-quadruplex DNA over duplex DNA. Spectroscopic and molecular docking studies suggest that the complexes 2–3 interact with telomeric G-quadruplex DNA mainly through groove binding. The compounds 1–3 are emissive (Φem > 0.22), making it possible to study the localization of 1–3 in A549 using fluorescence microscopy. The complexes 2–3 are mainly localized in nuclei, while 1 is localized in the nuclei and cytoplasmic region after 0.5 h incubation. The complex 3 inhibits A549 cells selectively over non-cancerous NIH3T3 cells, with higher antitumor activity than 1 and cisplatin.
 Please wait while we load your content...
Please wait while we load your content...

