
Ultra-rapid pattern formation of block copolymers with a high-χ parameter in immersion annealing induced by a homopolymer†
Myung Jin Kim‡
a,
Woon Ik Park‡b,
Young Joong Choic,
Yun Kyung Jungd and
Kwang Ho Kim*bc
aSchool of Convergence Science, Pusan National University, Geumjeong-gu, Busan 609-735, Republic of Korea
bGlobal Frontier R&D Center for Hybrid Interface Materials (HIM), Geumjeong-gu, Busan 609-735, Republic of Korea
cSchool of Materials Science and Engineering, Pusan National University, Geumjeong-gu, Busan 609-735, Republic of Korea. E-mail: kwhokim@pusan.ac.kr
dSchool of Natural Science, Ulsan National Institute of Science and Technology (UNIST), Ulju-gun Ulsan 689-798, South Korea
First published on 11th February 2016
Abstract
The directed self-assembly (DSA) of block copolymers (BCPs) has attracted considerable attention due to the outstanding ability of this method to complement or replace the conventional photolithography process. However, there are critical issues to resolve in order to realize the rapid pattern formation of BCPs with a high Flory–Huggins parameter (χ). Here, we introduce a simple method to expedite the self-assembly kinetics with the addition of a polystyrene homopolymer (hPS) to poly(styrene-b-dimethylsiloxane) (PS-b-PDMS) BCPs with a high-χ parameter. We provide a systematic presentation of how the hPS affects the self-assembly of PS-b-PDMS BCPs in the immersion annealing process. We found the optimum annealing conditions of the mixing ratio for the hPS/PS-b-PDMS BCP blends, showing a very short annealing time (<1 min) to obtain highly-ordered nanostructures. In addition, we discuss how the annealing temperature and mixing ratio of the binary solvent improve the self-assembly kinetics of the hPS/PS-b-PDMS BCP blends, suggesting a new route which effectively enhances the self-assembly speed. We believe that this facile and useful approach is applicable to the other BCP combination studies, contributing to the development of the next-generation BCP lithography.
1. Introduction
The directed self-assembly (DSA) of block copolymers (BCPs) has been known as one of the most promising candidates for next-generation lithography owing to the advantages of an excellent pattern resolution with a simple process, and good cost-effectiveness.1–8 The self-assembly of BCP, which consists of two or more mutually immiscible polymer blocks with covalent bonding at the end, can produce diverse morphologies with a size range of 5–50 nm, such as spheres, cylinders, perforated lamellae, and lamellae.5,9–20 Previous studies have shown many advantages of BCPs with a large Flory–Huggins interaction parameter (χ) along with the achievement of scaling to the sub-10 nm regime, when compared to optical lithography.18,21 The use of high-χ BCPs is especially critical to reduce the pattern size, pattern period, line edge roughness, and thermodynamic defect density, all of which can greatly decrease with the χ-parameter.22–24 On the other hand, the BCP chain mobility exponentially decreases in proportion to the χ-value; therefore, high-χ BCPs typically require solvent vapor annealing to produce well-ordered nanostructures to increase the polymer chain mobility effectively at lower temperatures compared to thermal annealing.25 Moreover, solvent vapor annealing can lead to excellent tunability of the geometry of self-assembled BCP patterns, showing variations of the morphology, microdomain size, and pattern period through control of the compositions of the solvent vapors.19–21In spite of such benefits of solvent vapor annealing, many research groups who study BCP have been to develop new annealing processes which improve the self-assembly kinetics of high-χ BCPs, as dozens of hours at room temperature are required to obtain well-organized nanostructures for high-χ BCPs by typical solvent vapor annealing.21,26 During the self-assembly process of BCPs, the polymer chain mobility or inter-diffusivity (D) exponentially decreases with the χ-value, which is expressed as D ∼ exp(−χNfAfB), where N is the number of repeating units, and fA and fB are the volume fractions of A-b-B BCP, respectively.25,27 To expedite the self-assembly of high-χ BCPs, thermally assisted solvent annealing methods were recently reported, including a microwave-assisted solvent annealing and solvothermal annealing.28–30 These annealing methods showed how the ultrafast pattern formation of well-ordered BCP can be achieved by accelerating the evaporation of the solvent vapors, demonstrating short annealing times of only a few minutes. Moreover, a mixed solvent annealing method was introduced, leading to both rapid self-assembly kinetics and excellent morphological tunability of high-χ BCPs using a binary solvent composed of two selective solvents for each block.31,32 These results commonly demonstrate methods which enhance the BCP self-assembly kinetics, suggesting useful and simple approaches to induce lower activation energy levels for chain diffusion.
In addition, we lately introduced a novel and practical immersion annealing method to realize rapid self-assembly, process simplicity, and easy controllability for high-χ BCPs.20 The simple immersion of BCP thin films in a solvent mixture consisting of a non-swelling solvent and a swelling solvent can induce the microphase separation of the BCPs, showing well-ordered nanostructures in a few minutes under composition-optimized solvent conditions. Moreover, the immersion of one BCP into the binary solvent leads to considerable morphological tunability, showing several different morphologies, such as spheres, cylinders, perforated lamellae, lamellae, core–shell spheres, core–shell cylinders, and sugarcane cylinders. However, if further improvement of the self-assembly kinetics during immersion annealing can be achieved, the high manufacturing cost due to the high throughput during the pattern formation process will be reduced significantly.
Here, we present a simple and cost-efficient way to promote the self-assembly of BCPs by employing homopolymer/high-χ BCP blends based on immersion annealing. For poly(styrene-b-dimethylsiloxane) (PS-b-PDMS) di-BCP with a high χ (∼0.26), we investigated how the mixing ratio of a polystyrene homopolymer (hPS) influences the self-assembly kinetics of BCPs to obtain well-aligned nanostructures, indicating composition-boundary conditions for microphase separation and macrophase separation. Furthermore, we successfully realized an extremely short annealing time (∼10 s) by controlling the volume fraction of the binary solvent (toluene and ethanol). We also demonstrated how ultra-rapid BCP pattern formation with good ordering can be accomplished in ten seconds by accelerating the annealing temperature.
2. Experimental
2.1. Self-assembly of the hPS/PS-b-PDMS BCP blend
The PS-b-PDMS BCP, PS homopolymer (hPS), and hydroxy-terminated PS homopolymer (PS–OH) were provided by Polymer Source Inc. of Canada. The Si substrates with trenches (width: 1 μm, depth: 40 nm) were fabricated by conventional photolithography and a dry etching process. The surfaces of the Si trench templates were modified by a PS–OH homopolymer with a MW of 38 kg mol−1 at 150 °C for about two hours in a vacuum oven, followed by washing with toluene to remove any unreacted PS residue. The sphere-forming PS-b-PDMS BCP (MW = 55 kg mol−1, fPDMS = 9.8%) and hPS (MW = 2.7 kg mol−1) were dissolved in toluene at 1.0 weight percent (wt%), after which the dissolved BCP solution and hPS solution were mixed at various mixing ratios. The blended polymer solutions were spin-coated at high speed (4000 rpm at room temperature) for 30 seconds onto the PS brush-treated Si substrates followed by immersion-annealed at 65 °C for 10 s to 30 min in a stainless steel chamber with a mixed solvent of toluene and ethanol. After the annealing process, all of the samples were treated with CF4 plasma (25 s at 50 W) followed by O2 plasma (25 s at 60 W) using a reactive ion etching (RIE) system, thus resulting in self-assembled SiOx or oxidized PDMS patterns.332.2. Scanning electron microscope (SEM)
All the self-assembled BCP or hPS/BCP microdomains were observed after the RIE process. The self-assembled PS/PS-b-PDMS blend morphologies were observed using a field emission scanning electron microscope (FE-SEM: Hitachi S-4800) which was operated with an acceleration voltage of 5–10 kV, an emission current of 5–10 μA, and a working distance of 4–8 mm.2.3. Film thickness measurement
The film thickness of hPS/BCP blend was measured using a reflectometry system (F20-UV, FILMETRICS Inc., USA). We at first measured the initial thickness of the immersion-annealed polymer film in the binary solvent which consists of swelling solvent (toluene) and non-swelling solvent (ethanol), and monitored the variation of the film thickness for 300 seconds during the immersion annealing process. After measuring the film thickness, the value was divided by initial thickness of BCP film, indicating the swelling ratio (SR) of each polymer film.3. Results and discussion
3.1. Conceptual illustration of the rapid self-assembly of the PS/PS-b-PDMS BCP blend
Fig. 1 and S1† show the experimental setup used for the immersion annealing process and a conceptual illustration of the rapid self-assembly kinetics of the hPS/PS-b-PDMS BCP blend. The hPS/PS-b-PDMS blend samples are immersed in a binary solvent which consists of a swelling solvent (toluene) and a non-swelling solvent (ethanol) in a stainless steel annealing chamber on a hot stage, showing the BCP thin film completely covered with the mixed solvent. During the immersion annealing process, while the non-swelling solvent does not cause any swelling of the BCP owing to the big difference of Hildebrand solubility parameter (δethanol ∼ 26.2 MPa1/2, δPS ∼ 18.5 MPa1/2, and δPDMS ∼ 15.5 MPa1/2), the swelling solvent (δtoluene ∼ 18.3 MPa1/2) induces microphase separation of the BCP thin film through the penetration of the molecules into the BCP thin film, resulting in a well-ordered BCP pattern. After O2 plasma treatment, the Si-containing PDMS block of PS-b-PDMS BCP is easily converted to oxidized PDMS (ox-PDMS, SiOx) patterns due to the high etch selectivity (>10) between the PS and the PDMS blocks, whereas the PS block is completely removed, as described in Fig. 1b. The addition of hPS to PS-b-PDMS (BCP blend) leads to faster pattern formation of the self-assembled BCP for a short annealing time compared to the PS-b-PDMS without any hPS. | ||
| Fig. 1 Schematic representation of the experimental setup for immersion annealing, and self-assembly of the PS/PS-b-PDMS BCP blend. (a) Structural formula of the poly(styrene-b-dimethylsiloxane) (PS-b-PDMS) and PS homopolymer (left), and experimental setup for immersion annealing (right). (b) Procedure for the rapid formation of a self-assembled PS/PS-b-PDMS pattern using immersion annealing. | ||
3.2. Microphase and macrophase separations of the PS/PS-b-PDMS BCP blend
First, we investigated how the addition of hPS to PS-b-PDMS (SD55, fPDMS = 9.8%) BCP with a MW of 55 kg mol−1 affects the swelling ratio (SR = swollen thickness divided by the initial thickness). Fig. 2 shows the SR measurement curves of the immersed BCP thin film in a binary solvent of toluene and ethanol with a mixing ratio (Vtol/Veth) of 0.43, depending on the mixing ratio of hPS with a MW of 2.7 kg mol−1 (0–30%). The graph on the right in Fig. 2 is a median plot of the SR curves on the left. The SR of the BCP thin film without the addition of hPS (0%, pure BCP) shows a relatively large value (∼1.1), resulting from the penetration of toluene (δtol = 18.3 MPa1/2) which is a good solvent for the majority PS block (fPS = 90.2%) into the SD55 BCP. The SR value increased in proportion to the mixing ratio of hPS in a range of 5–15%, whereas the SR value was decreased significantly to ∼0.8 due to the excessive addition of hPS (>20%) into the BCP. Based on previous studies, BCP with a homopolymer can be microphase-separated by the uniform solubilization of a homopolymer into the BCP domain, when the mixing ratio of the homopolymer in the hPS/BCP blend is appropriately low.34,35 The primary reason for the increase in SR is likely the result of decrease of the volume fraction of the minority PDMS block (fPDMS = 9.8%), as the inter-diffusivity decreases exponentially with the multiplication of two volume fractions (fA × fB), resulting in the rapid self-assembly of BCP. The second reason is likely the distribution of the PS chains, which may be more uniform due to the positioning of the hPS microdomain over the entire PS matrix. The uniformly distributed PS chains can be more swollen by PS-preferential toluene molecules, along with an increase of fPS or a decrease of fPDMS. However, when the mixing ratio of hPS exceeds 20%, the SR value decreases considerably, implying that toluene molecules can rather cause the damage or dewetting of BCP film owing to the significant increase of the effective volume fraction of the PS chains (feffPS). Here, we can expect that an excessive amount of hPS will bring about macrophase separation of the hPS/SD55 blend between hPS and SD55 BCP, showing damaged hPS regions after an immersion annealing process.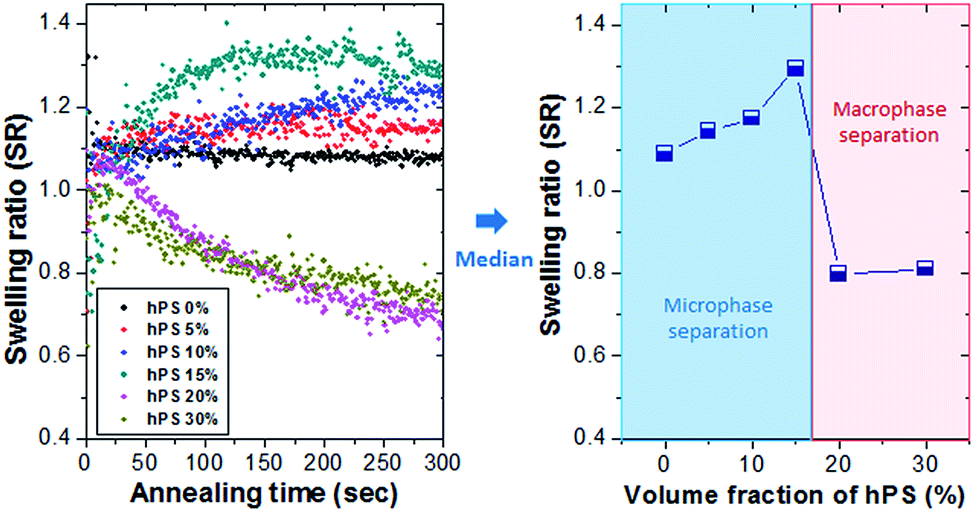 | ||
| Fig. 2 Measurement of the swelling ratios (SRs) of the SD55 BCP with/without hPS. (Left) Swelling ratios of the hPS/SD55 BCP blend and the pure SD55 BCP. (Right) Median plot of left graph. | ||
Fig. 3 and S2† show self-assembled SD55 dot patterns which were immersion-annealed with varied mixing ratios of hPS for one minute at a fixed Vtol/Veth ratio of 0.43. All of the scanning electron microscope (SEM) images show revealed oxidized PDMS or SiOx patterns after O2 plasma treatment. The inset images are fast Fourier transformation (FFT) patterns obtained from each SEM image, indicating the ordering of the self-assembled BCP patterns. As expected, the ordering of the hPS/SD55 BCP pattern was improved depending on the mixing ratio of hPS. As the mixing ratio of hPS into BCP increases, the ordering of self-assembled nanostructures increases in the range of 0–15%, coinciding with the SR measurement results (Fig. 2). The SD55 BCP with the appropriate mixing ratios of hPS (5–15%) induces rapid self-assembly kinetics to obtain a highly ordered dot pattern. On the other hand, when the mixing ratio of hPS exceeds 15% (20–50%), the BCP patterns showed both microphase-separated SD55 and isolated domains, implying that a large amount of homopolymer can be separated from self-assembled BCP. It is likely that a high concentration of a homopolymer cannot be fully solubilized in the BCP. Accordingly, the islands are presumed to be hPS domains damaged by toluene molecules or macrophase-separated hPS regions removed by O2 plasma, suggesting a boundary between the microphase and macrophase separations in the hPS/SD55 BCP blend.
 | ||
| Fig. 3 Self-assembled morphologies of the SD55 BCP with/without hPS. Self-assembled dot patterns of the hPS/SD55 blend depending on the mixing ratio of hPS at a fixed Vtol/Veth ratio of 0.43 for one minute. The inset images show the fast Fourier transforms (FFTs), indicating a well-ordered dot pattern. The SD55 BCP with the appropriate mixing ratios of hPS (15%) produces a highly ordered dot pattern in one minute, an excessive addition of the hPS will cause macrophase separation of the hPS/SD55 blend between the hPS and the SD55 BCP. | ||
3.3. Self-assembly kinetics dependent on the mixing ratio of hPS into PS-b-PDMS
Fig. 4 shows the annealing time required to obtain highly ordered SD55 patterns, indicating the dependency on the mixing ratio of hPS (0–15%) at a fixed Vtol/Veth ratio of 0.43. The hPS (15%)/SD55 BCP blend can produce a well-ordered dot pattern after a short immersion time of 30 s, which is ten times faster compared to the pure SD55 BCP (hPS 0%, 300 s). The diameter (dot size) and period (center-to-center distance) of the self-assembled SD55 pattern were approximately 16 nm and 35 nm, respectively. They did not indicate prominent changes when the mixing ratio of the hPS was varied. It is likely that a hPS with a small MW (2.7 kg mol−1) can be well solubilized in the SD55 BCP with a relatively large MW (55 kg mol−1). Fig. S3 in the ESI† shows the time-evolution to obtain well-ordered nanostructures for mixing ratios of hPS (0% & 15%) when the Vtol/Veth ratio is 0.43. These results strongly suggest that the addition of a homopolymer into the BCP within a suitable volume fraction range can induce rapid pattern formation of BCPs with good ordering. | ||
| Fig. 4 Self-assembled dot patterns depending on the volume fraction of hPS when Vtol/Veth = 0.43. (a) Graph for the annealing time to obtain a well-ordered dot pattern, and pattern periods and dot sizes vs. the mixing ratio of hPS. (b) Annealing times required to obtain highly ordered nanostructures, indicating the dependency on the mixing ratio of hPS (0–15%). The hPS (15%)/SD55 BCP blend can generate a well-ordered dot pattern in 30 s, which is ten times faster than pure SD55 (hPS 0%, 300 s). | ||
3.4. Ultra-rapid pattern formation of the hPS (15%)/SD55 BCP blend
Based on our previously reported studies, the self-assembly speed of the hPS/SD55 BCP blend can be further boosted by increasing the mixing ratio of toluene to ethanol from 0.43 to 0.67, as shown in Fig. 5.20 While the non-swelling solvent of ethanol does not affect the self-assembly of the hPS/SD55 BCP blend, the amount of the swelling solvent (toluene) influences the self-assembly kinetics and is thus a critical factor determining the annealing time to obtain well-ordered nanostructures, as described above. The graph in Fig. 5a shows the optimum annealing time for well-ordered dot patterns of SD55 BCP with/without hPS (0% & 15%) depending on the Vtol/Veth ratio. The annealing time decreases in proportion to the Vtol/Veth ratio, showing faster self-assembly kinetics of the hPS/SD55 BCP blend compared to the pure SD55, while a similar tendency for the generation of highly ordered dot patterns was observed between the hPS/BCP blend and the pure BCP. While the same annealing time between two samples was required when the Vtol/Veth ratio was 0.25, the gap in the annealing time increased when the Vtol/Veth ratio was 0.67. The diameter/period of the self-assembled hPS/SD55 BCP pattern slightly increased from 15.8/33.2 nm when the Vtol/Veth was 0.25 to 16.7/35.7 nm when Vtol/Veth = 0.67, as shown in Fig. 5b. Fig. 5c and S4† show the ordered dot patterns under the optimum annealing conditions (with the annealing time at different Vtol/Veth ratios) for the hPS/SD55 blend and the pure SD55. These results may stem from the higher swelling of the hPS/SD55 blend due to the much more toluene vapors when the Vtol/Veth ratio is 0.67 compared to when Vtol/Veth = 0.25, showing a similar effect of the solvent vapor pressure during the solvent vapor annealing process. | ||
| Fig. 5 Ultra-rapid pattern formation of the hPS (15%)/SD55 BCP blend by controlling the mixing ratio of binary solvent (Vtol/Veth ratio). (a) Optimum annealing time to obtain well-ordered dot patterns of SD55 BCP with/without hPS (0% & 15%) depending on the Vtol/Veth ratio. (b) Graph for dot size/period vs. Vtol/Veth ratio of the self-assembled nanostructure. (c) Well-ordered dot patterns of pure SD55 and hPS (15%)/SD55 blend immersion-annealed for 3 min and 10 s, respectively, when Vtol/Veth = 0.67. | ||
Furthermore, we successfully realized an extremely short annealing time (∼10 s) to obtain a highly organized dot pattern of the hPS (15%)/SD55 BCP blend by increasing the annealing temperature. Fig. 6a shows the ordered dot pattern of the hPS (15%)/SD55 BCP blend immersion-annealed when Vtol/Veth = 0.43 at annealing temperatures of 65 °C, 75 °C, and 85 °C, showing short annealing times of 30 s, 20 s, and 10 s, respectively. The diameter/period of the self-assembled hPS/SD55 BCP pattern increased slightly in proportion to the annealing temperature, as shown in Fig. 6b. Ultra-rapid pattern formation at 85 °C may be attributed to the higher swelling of the hPS/SD55 BCP thin film caused by the fast mobility of toluene molecules at a higher temperature, showing similarity with solvothermal annealing. Here, it should be noted that more rapid pattern formation of high-χ BCPs can be realized by controlling the annealing temperature in conjunction with the mixing ratio of the binary solvent.
 | ||
| Fig. 6 Thermally accelerated fast pattern formation of the hPS (15%)/SD55 BCP blend. (a) Highly ordered dot patterns at different annealing temperatures when Vtol/Veth = 0.43. (b) Graph for annealing time, and dot size/period vs. annealing temperature. | ||
4. Conclusions
In summary, we introduced a facile and useful approach to expedite the self-assembly of BCP by employing homopolymer/high-χ BCP blends based on the immersion annealing. For PS-b-PDMS with a high-χ (∼0.26), rapid pattern formation of BCP was achieved by the appropriate addition of a PS homopolymer into PS-b-PDMS BCP, showing a short annealing time of 30 seconds. For the hPS/PS-b-PDMS blend, we successfully achieved a highly ordered dot pattern with an extremely short immersion time of 10 seconds by controlling the mixing ratio of the binary solvent. Furthermore, we demonstrated how ultra-rapid BCP pattern formation with good ordering can be accomplished within 10 seconds by accelerating the annealing temperature, suggesting a new route which effectively enhances the self-assembly speed. We believe that this simple and practical methodology may be widely applicable for the rapid self-assembly of other BCPs, contributing to the next-generation BCP lithography.Acknowledgements
This research was supported by Global Frontier Program through the Global Frontier Hybrid Interface Materials (GFHIM) of the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (No. 2013M3A6B1078874).Notes and references
- T. L. Morkved, M. Lu, A. M. Urbas, E. E. Ehrichs, H. M. Jaeger, P. Mansky and T. P. Russell, Science, 1996, 273, 931–933 CAS.
- T. Thurn-Albrecht, J. Schotter, C. A. Kastle, N. Emley, T. Shibauchi, L. Krusin-Elbaum, K. Guarini, C. T. Black, M. T. Tuominen and T. P. Russell, Science, 2000, 290, 2126–2129 CrossRef CAS PubMed.
- S. O. Kim, H. H. Solak, M. P. Stoykovich, N. J. Ferrier, J. J. de Pablo and P. F. Nealey, Nature, 2003, 424, 411–414 CrossRef CAS PubMed.
- M. P. Stoykovich, M. Muller, S. O. Kim, H. H. Solak, E. W. Edwards, J. J. de Pablo and P. F. Nealey, Science, 2005, 308, 1442–1446 CrossRef CAS PubMed.
- I. Bita, J. K. W. Yang, Y. S. Jung, C. A. Ross, E. L. Thomas and K. K. Berggren, Science, 2008, 321, 939–943 CrossRef CAS PubMed.
- M. P. Stoykovich and P. F. Nealey, Mater. Today, 2006, 9, 20–29 CrossRef CAS.
- R. A. Segalman, Science, 2008, 321, 919–920 CrossRef CAS PubMed.
- S. J. Jeong, J. Y. Kim, B. H. Kim, H. S. Moon and S. O. Kim, Mater. Today, 2013, 16, 468–476 CrossRef CAS.
- J. Y. Cheng, A. M. Mayes and C. A. Ross, Nat. Mater., 2004, 3, 823–828 CrossRef CAS PubMed.
- R. Ruiz, H. M. Kang, F. A. Detcheverry, E. Dobisz, D. S. Kercher, T. R. Albrecht, J. J. de Pablo and P. F. Nealey, Science, 2008, 321, 936–939 CrossRef CAS PubMed.
- R. A. Segalman, H. Yokoyama and E. J. Kramer, Adv. Mater., 2001, 13, 1152–1155 CrossRef CAS.
- C. T. Black, R. Ruiz, G. Breyta, J. Y. Cheng, M. E. Colburn, K. W. Guarini, H. C. Kim and Y. Zhang, IBM J. Res. Dev., 2007, 51, 605–633 CrossRef CAS.
- J. Chai, D. Wang, X. N. Fan and J. M. Buriak, Nat. Nanotechnol., 2007, 2, 500–506 CrossRef PubMed.
- S. B. Darling, Prog. Polym. Sci., 2007, 32, 1152–1204 CrossRef CAS.
- C. M. Bates, T. Seshimo, M. J. Maher, W. J. Durand, J. D. Cushen, L. M. Dean, G. Blachut, C. J. Ellison and C. G. Willson, Science, 2012, 338, 775–779 CrossRef CAS PubMed.
- K. G. A. Tavakkoli, K. W. Gotrik, A. F. Hannon, A. Alexander-Katz, C. A. Ross and K. K. Berggren, Science, 2012, 336, 1294–1298 CrossRef CAS PubMed.
- C. A. Ross, K. K. Berggren, J. Y. Cheng, Y. S. Jung and J. B. Chang, Adv. Mater., 2014, 26, 4386–4396 CrossRef CAS PubMed.
- J. W. Jeong, W. I. Park, M. J. Kim, C. A. Ross and Y. S. Jung, Nano Lett., 2011, 11, 4095–4101 CrossRef CAS PubMed.
- W. I. Park, Y. Kim, J. W. Jeong, K. Kim, J. K. Yoo, Y. H. Hur, J. M. Kim, E. L. Thomas, A. Alexander-Katz and Y. S. Jung, Sci. Rep., 2013, 3, 3190 Search PubMed.
- W. I. Park, J. M. Kim, J. W. Jeong and Y. S. Jung, ACS Nano, 2014, 8, 10009–10018 CrossRef CAS PubMed.
- Y. S. Jung and C. A. Ross, Adv. Mater., 2009, 21, 2540–2545 CrossRef CAS.
- S. Park, D. H. Lee, J. Xu, B. Kim, S. W. Hong, U. Jeong, T. Xu and T. P. Russell, Science, 2009, 323, 1030–1033 CrossRef CAS PubMed.
- J. M. Kim, Y. J. Kim, W. I. Park, Y. H. Hur, J. W. Jeong, D. M. Sim, K. M. Baek, J. H. Lee, M. J. Kim and Y. S. Jung, Adv. Funct. Mater., 2015, 25, 306–315 CrossRef CAS.
- P. N. Patrone and G. M. Gallatin, Macromolecules, 2012, 45, 9507–9516 CrossRef CAS.
- T. P. Lodge and M. C. Dalvi, Phys. Rev. Lett., 1995, 75, 657–660 CrossRef CAS PubMed.
- S. H. Kim, M. J. Misner, T. Xu, M. Kimura and T. P. Russell, Adv. Mater., 2004, 16, 226–231 CrossRef CAS.
- R. Ruiz, J. K. Bosworth and C. T. Black, Phys. Rev. B: Solid State, 2008, 77, 054204 CrossRef.
- X. Zhang, K. D. Harris, N. L. Wu, J. N. Murphy and J. M. Buriak, ACS Nano, 2010, 4, 7021–7029 CrossRef CAS PubMed.
- W. I. Park, K. Kim, H. I. Jang, J. W. Jeong, J. M. Kim, J. Choi, J. H. Park and Y. S. Jung, Small, 2012, 8, 3762–3768 CrossRef CAS PubMed.
- K. W. Gotrik and C. A. Ross, Nano Lett., 2013, 13, 5117–5122 CrossRef CAS PubMed.
- W. I. Park, S. Tong, Y. Liu, I. W. Jung, A. Roelofs and S. Hong, Nanoscale, 2014, 6, 15216–15221 RSC.
- W. I. Park, Y. J. Choi, J. M. Yun, S. W. Hong, Y. S. Jung and K. H. Kim, ACS Appl. Mater. Interfaces, 2015, 7, 25843–25850 CAS.
- W. I. Park, J. M. Yoon, M. Park, J. Lee, S. K. Kim, J. W. Jeong, K. Kim, H. Y. Jeong, S. Jeon, K. S. No, J. Y. Lee and Y. S. Jung, Nano Lett., 2012, 12, 1235–1240 CrossRef CAS PubMed.
- H. K. Lee, C. K. Kang and W. C. Zin, Polymer, 1996, 37, 287–295 CrossRef CAS.
- X. A. Li, L. J. Xue and Y. C. Han, J. Mater. Chem., 2011, 21, 5817–5826 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra00350h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |