
Bioengineering three-dimensional culture model of human lung cancer cells: an improved tool for screening EGFR targeted inhibitors
Dan-Dan Wang†
a,
Wei Liu†b,
Jing-Jie Changb,
Xu Chengb,
Xiu-Zhen Zhangb,
Hong Xub,
Di Fengb,
Li-Jun Yu*b and
Xiu-Li Wang*b
aDalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
bCollege of Basic Medical Science, Dalian Medical University, Dalian 116044, China. E-mail: panpan1210@dicp.ac.cn; yulijundl1963@163.com
First published on 26th February 2016
Abstract
Cell models have been widely accepted to explore the mechanisms of tumorigenesis and predict drug response. Unfortunately, there is still a lack of accurate models that can highly recapitulate the important molecular events occurring in the human non-small-cell lung cancer patient's body. We hypothesize that the 3D microenvironment provided by collagen hydrogel would not only promote generation of tumor cultures that more closely resemble the in vivo tumor morphology, but also contribute to bioengineering a well-defined non-small-cell lung cancer culture model that potentially enhances the molecularly targeted drug evaluation. Here, we developed a more pathologically relevant, three-dimensional culture model of human lung cancer cells through embedding A549 cells within a collagen hydrogel. Our results showed that the collagen-embedded A549 cells formed numbers of cell aggregates that displayed good viability and more tissue-like morphology. A significantly increased expression level of EGF/EGFR was also observed in the 3D cultures. More importantly, the 3D culture had the ability to respond to treatment by the targeted inhibitor, as evidenced by its enhanced drug resistance and inhibited growth. Thus, this in vitro culture system effectively represents an improved culture model of human lung cancer and would potentially provide a robust tool for mechanism-based anti-lung cancer drug discovery and development.
Introduction
Non-small-cell lung cancer (NSCLC) is one of the leading causes of cancer-related deaths in the world. To date, no effective therapy in clinic has been established to significantly improve the overall 5 year survival rate of NSCLC patients. This may be mainly due to the lack of more accurate models that could highly recapitulate the important biological and molecular events that occur in the NSCLC patient's body. Cell models have been widely accepted to explore the mechanisms of tumorigenesis and predict drug response. Monolayer-based cell culture assays have been widely accepted to explore the mechanisms of tumorigenesis and metastasis, identify cancer biomarkers and even predict their drug response. However, this conventional two-dimensional (2D) culture system has recently been found to be deficient in mimicking the in vitro tumor microenvironment more closely. This is important because an increasingly number of investigations have supported that tumor microenvironment (TM), which is collectively composed of stromal cells, extracellular matrix (ECM), soluble growth factors and as well as cell–cell or cell–ECM interactions, plays central role in determining the behavior of cancer cells.1–5 Animal models to some extent provide in vivo microenvironment for some particular pathological processes, however, due to their complicated conditions and species differences, there is often a lack of correlation between animal experiments and human trials. This is specifically more evident in their response to drug therapy, accounting for the higher failure rate (approximately 95% for anti-cancer drug discovery).6 As a result, developing an improved tumor model that capable of replicating the local TM to the extent possible, including all the key factors and their complex interactions, has become one of the most critical challenges in cancer research.In this regard, three-dimensional (3D) cultures have increasingly become more attractable owing to their great potential of mimicking the native tissue microenvironment in vitro. Compared to the 2D monolayer cultures, 3D culture systems not only provide 3D architectural space for cell growth, but also incorporate ECM and/or stromal cells contributing to recapitulation of cell-matrix/cell interactions.7–9 A significantly distinct gene expression profile has been found between the 2D culture and the 3D culture of several cell lines, further supporting the profound influence exerted by the culture microenvironment.10–12 Nowadays, lots of advances have been reported in establishing 3D cell culture in vitro. However, to date, none of accurate models that could highly recapitulate the important molecular events occurring in the human non-small cell lung cancer patient's body have been reported yet.
In the present study, we described a 3D culture system of NSCLC by embedding the immortalized A549 cells within collagen gel. We hypothesize that the collagen hydrogel is capable of providing a 3D microenvironment that promotes more tumor-like culture formation in vitro, recapitulating the tumor cells' phenotype and activity in vivo more closely. And this might further contribute to generating a more defined NSCLC culture model that potentially enhances the molecularly targeted drug evaluation. Towards this goal, the constructed 3D culture model of A549 cells was characterized by its growth profile, histology and gene expression. Moreover, its capability of responding to the targeted medicine was also evaluated through gefitinib treatment. It is expected that this 3D culture system would provide an improved model to better explore the pathophysiology of tumor cells or even aid in the discovery of novel anticancer therapies.
Results
Growth profile of A549 cells in collagen gel
Compared to the adherently, short spindle-shaped A549 cells in monolayer culture, a number of cellular spheroids were generated after 4 days of cultivation in collagen gel. These cell spheroids were found to be evenly distributed in the 3D architectural culture space, so that it's hard to be focused when their images were captured under the phase contrast microscope. Moreover, the size of these cell spheroids increased progressively over time, indicating their cellular proliferation during the culture in vitro (Fig. 1a1 and a2). | ||
| Fig. 1 Growth profile and viability of A549 cells within collagen hydrogel (at day 6 and day 12). Morphology images showed morphological characteristics of the structures formed by A549 cells in collagen hydrogel (a1 & a2). Viability of A549 cells in different groups detected by live/dead staining (green, live cells, (b1 & b2)). | ||
Viability of A549 cells embedded in collagen gel was also assessed by calcein-AM/EthD-1 staining. After 12 days of cultivation, the majority of cells involved by the cellular spheroids exhibited good viability (Fig. 1b1 and b2), suggesting that the 3D collagen matrix could provide a suitable microenvironment for the growth of A549 cells.
Proliferation of A549 cells in collagen gel
Proliferation of A549 cells under 3D culture condition was determined through DNA content assay. As shown in Fig. 2, DNA content of the A549 cells in collagen gel increased progressively during 11 days of cultivation. A similar growth trend of cells was also observed in the monocultures during the initial 5 days of culture in vitro. However, its DNA content (the monolayer cultures) reached a peak level quickly after 5 days of culture, which is much earlier than that of the 3D cultures. In addition, a noticeable increase of cell number in the 3D cultures was observed from day 5 to day 11, showing a significantly enhanced proliferation rate of A549 cells in collagen gel when compared with that of the monolayer cultures. This suggests the capabilities of the collagen gel in supporting the growth of A549 cells within the 3D compartment. | ||
| Fig. 2 Proliferation kinetics of A549 cells in 2D and 3D culture at indicated time points by DNA quantification analysis. A549 cells in 3D culture proliferated progressively during 11 day cultivation, while the cell growth of monoculture reached a peak level quickly and decreased gradually afterward. | ||
Morphological characteristics of the 3D culture
A detailed morphological characteristic of A549 cells in collagen gel was revealed by H&E staining. As shown in Fig. 3, several cell aggregates were observed in the H&E stained sections, and their size increased gradually during the culture, which was consistent with what we observed under the phase-contrast microscopy. But by the end of 20 days of cultivation in collagen gel, central necrosis was found within some larger cell aggregates evidenced by some acinus-like structure formation (Fig. 3a3). This might be due to the poor transport of oxygen in the central part of the enlarged cell spheroids. Taking into the account the effects of culture time on the grow profile of A549 cells in collagen gel, we therefore determined to harvest the samples by the end of 12 days of cultivation for the following detection in order to achieve a better quality control.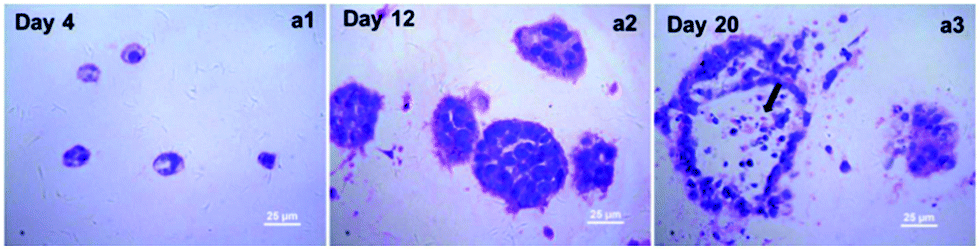 | ||
| Fig. 3 Morphological characteristics of A549 cells in 3D culture. H&E staining showed A549 cells formed aggregates and their size increased gradually during the culture. Center necrosis was found within some larger cell aggregates at day 20 (arrowhead notes center necrosis). | ||
SEM images further revealed the ultra-structure of the collagen fibers and the A549 cells embedded within the collagen hydrogel. As shown in Fig. 4, A549 cells were located in direct association with collagen fibers and successfully generated cell aggregate within the 3D collagen scaffolds. 12 days of cultivation confirmed our collagen-based 3D culture systems are able to attract and to support a cluster growth pattern of A549 cells.
 | ||
| Fig. 4 SEM images showed the collagen meshwork and A549 cell aggregates (noted by the arrow head) formed during the 3D cultivation in vitro. | ||
EGF/EGFR expression in the 3D culture
An increased transcript expression level of EGF and EGFR gene in the 3D cultures was detected out by qRT-PCR analysis when compared with that of the monolayer culture (Fig. 5). In particular, more than 350-fold higher expression of EGF gene was observed in the 3D culture. Accordingly, gene expression of EGFR was also improved. This data suggested profound influence exerted by the three-dimensional microenvironment on the gene expression of A549 cells.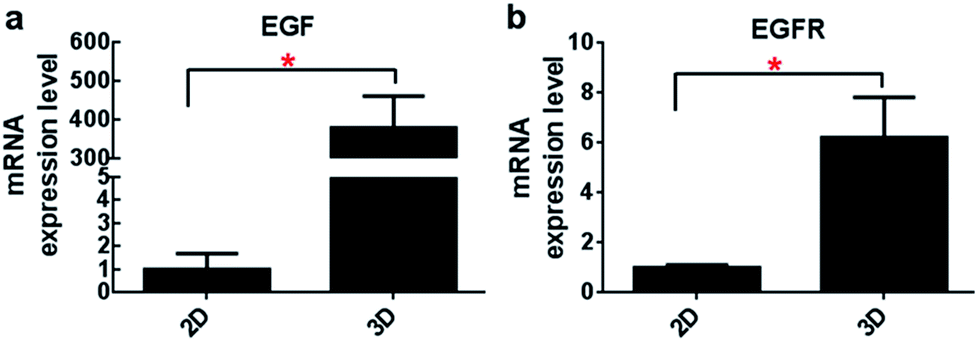 | ||
| Fig. 5 Transcript expression levels of EGF and EGFR by real-time RT-PCR at the indicated time points. A significant increase in the expression of both EGF and EGFR was detected in A549 cells culture in collagen hydrogel (n = 3, *p < 0.05). | ||
Compared to the monolayer culture, the content of EGF protein secreted by the 3D cultures (221.4 ± 13.8 pg per ml per day) was also found to be improved significantly evidenced by their nearly 18.3 times higher secretion of EGF detected by ELISA (12.1 ± 3.4 pg per ml per day in 2D culture, Fig. 6). This result was in accordance with their EGF gene expression level as described above, supporting the effects of 3D microenvironment on the cells' secretion activity.
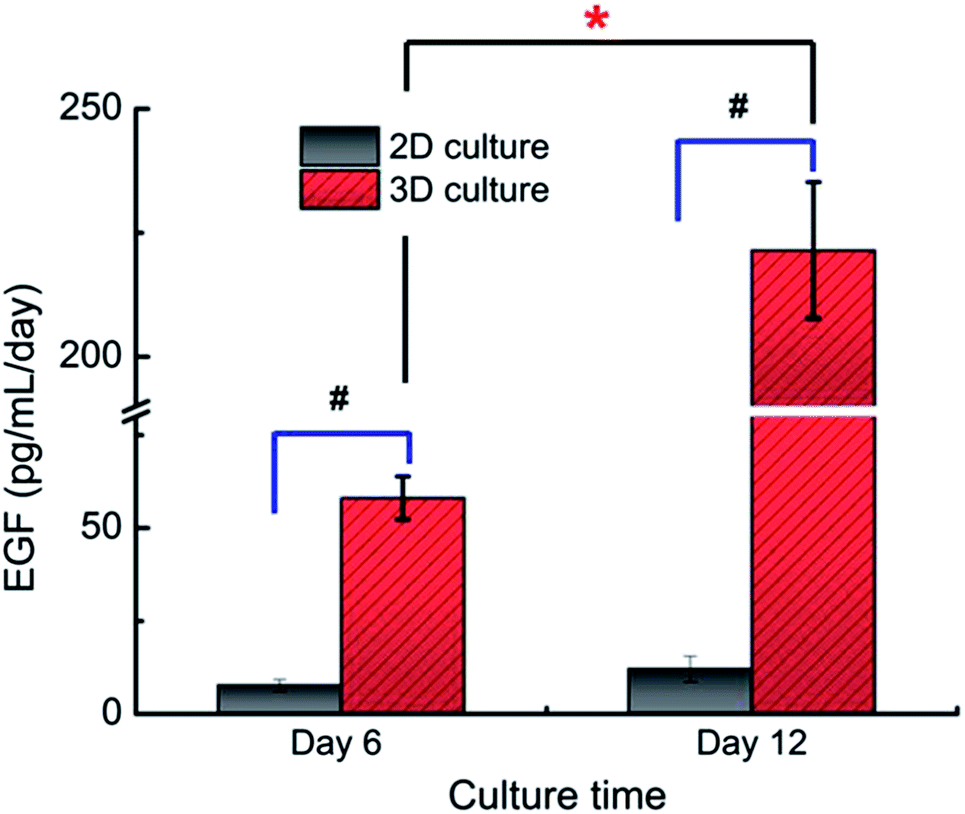 | ||
| Fig. 6 EGF secretion by A549 cells under different culture conditions assayed by ELISA. The content of EGF protein secreted by the 3D cultured A549 cells was significantly higher than that of the 2D culture (n = 3, #p < 0.05). The content of EGF protein secreted by the 3D cultured A549 cells at day 12 was significantly higher than that of day 6 (n = 3, *p < 0.05). | ||
Protein expression of EGFR, a trans-membrane receptor tyrosine kinase was also detected by immunofluorescence staining. As shown in Fig. 7a, more intensely staining of EGFR was found in the 3D cultures compared to the monolayer culture, suggesting an improved expression of EGFR in the A549 cell aggregates formed within 3D collagen gel. In agreement with this data, semi-quantitative analysis of western blotting further revealed a nearly 2.46 times higher expression of pho-EGFR (Tyr1092) in the 3D culture over the monolayer culture (*p < 0.001) (Fig. 7b). Pho-EGFR (Tyr1092) is an important form of active EGFR. This result further suggested that the unique culture microenvironment provided by 3D collagen gel was able to induce more EGFR phosphorylation in A549 cells, triggering downstream EGFR signaling transduction pathways to regulate cellular behavior.
 | ||
| Fig. 7 Protein expression of EGFR in the 2D and 3D cultures detected by immunofluorescence staining (a) and western blot (b). Cell nuclei were counterstained with DAPI (blue) and a higher EGFR expression level was observed in 3D culture (a). Consist with immunofluorescence staining, the semi-quantitative analysis of western blotting also showed a higher expression of pho-EGFR (Tyr1092) in the 3D culture over the monolayer culture (*p < 0.001) (b). | ||
Cytotoxicity and growth inhibitory effect of gefitinib on the 3D culture
We next examined the cytotoxicity effect of gefitinib, an EGFR targeting drug in the 3D cultures using AlamarBlue assay. As shown in Fig. 8a, the IC50 values of gefitinib were significantly enhanced in the 3D culture of A549 cells when compared to the monolayer culture. The difference of the IC50 values between them was up to 2.35 folds (127.89 μM vs. 54.32 μM), indicating the distinct drug responsive capability of A549 cells when they were cultured under different conditions. | ||
| Fig. 8 The cytotoxicity regression curve of A549 cells in 3D and 2D culture (a). The growth inhibitory curve of the 3D cultures with gefitinib treatment (b). Compared to the monolayer culture, the IC50 values of gefitinib were significantly enhanced in the 3D culture of A549 cells (a). This inhibitory effects of gefitinib on the 3D cultures displayed both dose- and time-dependent manner (b1) and the time point of drug supplementation also influenced the culture's response capability (b2). | ||
Given the fact that the traditional cytotoxic assay presents only an end-point evaluation for anti-cancer compounds, however, this may not be appropriate for EGFR targeted inhibitors because their mechanism of action is anti-proliferation without triggering apoptosis. Therefore, the growth inhibitory assay based on growth curve of the 3D cultures treated by gefitinib seems more suitable in order to obtain more detailed information of cells to the drug treatment or their resistance. Growth inhibitory effect of gefitinib on the 3D culture was observed as evidenced by the decreased DNA content after gefitinib treatment. This inhibitory effect displayed both dose- and time-dependent manner (Fig. 8b1). In addition, the time point of drug supplementation also influenced the culture's response capability. Compared to the 3D culture treated by the same concentration of gefitinib (50 μM and 100 μM) from day 7, more significant inhibition effect was observed in the 3D culture treated by gefitinib from day 1 (Fig. 8b2). This might be explained by the larger cell aggregates formation after 7 days and the different time length of the drug treatment.
Inhibition of pho-EGFR in the 3D culture
To further evaluate the drug response capability of the constructed 3D cultures, their pho-EGFR expression level after gefitinib treatment was also examined using western-blotting assay. As shown in Fig. 9, a remarkably decreased level of pho-EGFR was detected out in the 3D cultures treated by different concentration of gefitinib (25, 50, 100 μM). Similar to the growth inhibitory effect exhibited by gefitinib treatment, the inhibition of pho-EGFR was also displayed a concentration-dependent manner, which further support the drug responsive capability of the 3D cultures of A549 cells. | ||
| Fig. 9 Inhibition of pho-EGFR expression by gefitinib in the 3D culture using western-blotting assay. Gefitinib decreased the expression level of pho-EGFR in a dose-dependent manner in the 3D cultures (*p < 0.05, compare with 3D cultures with 0 μM of gefitinib treatment). | ||
Discussion
Cells cultivated within a 3D microenvironment in vitro are able to acquired phenotype and molecular signatures that closely mimics the in vivo biological system.13,14 This technology has been utilized in the field of tissue engineering and could also be potentially applied to construct a pathologically relevant tumor model in vitro (e.g. tumor engineering).15 Tumor engineering is defined as “the construction of complex culture models that recapitulate aspects of the in vivo tumor microenvironment to study the dynamics of tumor initiation, development, progression, and therapy on multiple scales”.16 A significant body of work in recent years has been devoted to development and characterization of 3D cultures,17 and the advantages of using 3D scaffold over 2D cell culture flask for acquiring an in vivo phenotype or function have also been documented.18–20 For example, multi-cellular spheroid-based culture of cancer cells has been found to have the ability to recreate the regions of differential growth/metabolism such as highly dividing cell at the surface, quiescent cell in the middle, and hypoxic necrotic cells at the center.21,22 This is important because anti-cancer drug resistance is readily expressed in the areas of poor diffusion and reduced vascularization. Our previous study on the encapsulated breast cancer cells (by using alginate-poly-lysine-alginate (APA) microcapsules) also supported an improved phenotypical stability of the cellular aggregated and their decreased drug resistance that more closely resembles to in vivo cancers.23Many advances have been reported in the past decades in establishing 3D cell culture in vitro, however, substantial progress still needs to be made before 3D tumor models become fully competent for replacing the traditional 2D cultures or even small animal models.24 It has been known that cells in vivo are either embedded within the ECM or in contact with the basement membrane (BM) during maintenance of normal morphology and function, and these cellular surroundings and the matrix components must be taken into account when constructing 3D tumor models because they also present in the tumor environment and play vital roles in determining tumor cell behavior.14,17,25,26 In the present study, we hypothesized that collagen hydrogel could be used as 3D culture scaffolds for bioengineering lung cancer models that mimic some key characteristics of in vivo NSCLC development. The reason that collagen hydrogel was selected in the present study is mainly due to its natural biocompatibility and 3D architecture of hydrogel that suitable for recreating the naturally hydrated microenvironment of solid tumor.27,28 In addition, collagen hydrogel can be tailored to stimulate some particular cellular responses within the constructed microenvironment.
And our study showed that through embedding A549 cells into collagen gel, the cells embedded were able to proliferate and gradually generate cell spheroids after 6 days of cultivation (Fig. 1a). These cell spheroids displayed good viability and more tissue-like morphology evidenced by their HE staining images and SEM images (Fig. 1b and 4). Representative necrosis in the central part of the cell spheroids was also observed after >20 days of culture in vitro (Fig. 3). This might be explained by the poor oxygen/nutrient availability and diffusion limitations within the larger cell spheroids. These characteristics performed by the 3D cultures of A549 cells somewhat recapitulate the developmental process of tumor in vivo that from single cell to multi-cell spheroid. In addition, this simple 3D culture of A549 cells exhibited significantly enhanced expression of EGF/EGFR genes and EGF/pho-EGFR proteins (Fig. 5–7). It has been well demonstrated that EGF/EGFR takes part in the process of pathogenesis and progression of several types of carcinomas, and the activation of EGFR tyrosine kinases plays central role in triggering downstream tumor biological event.29,30 So far the related mechanisms underlying the oncogenic potential of EGF/EGFR have been elucidated, which makes it become one of most promising therapeutic targets for anti-cancer therapy. Here, the mechanism of the enhanced expression of EGF/EGFR in the cell spheroids is still unknown, but we analyzed that collagen fibers in the hydrogel might exert some chemical, physical or even biological cues to influence the embedded A549 cells. Integrins, transmembrane receptors that are the bridges for cell–cell and cell–ECM interactions might be involved during this process. Specifically, an incorporation of collagen, one type of ligand of integrins, into the culture system could trigger integrins and their downstream chemical pathway to the interior of cells, resulting in a response such as regulation of gene transcription, cell proliferation.27 Several literatures have been published to support our analysis. For example, one study by Kiefer and Farach-Carson have reported that collagen type I affected the gene expression of prostate cancer cells through its interaction with α2β1 and α3β1 integrins, and facilitated the colonization and growth of metastatic of cancer cells.31 Paszek MJ and his colleagues also found that an integrated mechano regulatory circuit linking ECM to cytoskeleton through integrins was able to exert profound influence on the gene expression of some key signal molecules and regulate the related tissue phenotype.32
Considering the significantly enhanced expression of EGF/EGFR genes/proteins in the 3D culture of A549 cells, their drug response capability to the EGFR targeted inhibitor (gefitinib) was further evaluated. This is important because it might not only provide more information about the function of the 3D cultures, but also contribute to screen out those EGFR targeted compounds with higher efficiency and even better understanding mechanism of those drug candidates. Our results showed that compared to the 2D monolayer culture, the 3D culture of A549 cells displayed distinct drug sensitivity to gefitinib treatment evidenced by the increased IC50 value (Fig. 8a). This phenomenon is somewhat consistent with other previous reports in which 3D tumor cultures usually exhibited improved resistance in comparison with the monolayer cultures.33–35 This drug resistance might be caused by a decreased penetration of anti-cancer drugs, increased pro-survival signaling, and/or up-regulation of genes conferring drug resistance.36–38 In addition, integrin signaling pathway might also involve into this process to facilitate production of drug resistance within 3D microenvironment.
But it should be noted that as a targeted inhibitor, the clinically therapeutic effect of gefitinib is not cytotoxicity but growth inhibition through inhibiting EGFR activation. Thereafter, it seems that the end-point cytotoxicity assay (IC50 value) is not the most suitable approach to better evaluate the 3D cultures response to gefitinib treatment. To address this issue, the growth curves of 3D cultures (up to 14 days) with different concentration of gefitinib supplement were further detected in this study. And we found that the cell proliferation in the 3D cultures was significantly inhibited evidenced by the decreased DNA content after gefitinib treatment (Fig. 8b). This inhibition effect was both dose- and time-dependent, suggesting their capability in response to gefitinib treatment. Consistent with this result, a dose-dependent inhibition of pho-EGFR expression by gefitinib was also observed in the treated 3D cultures, which further supports their drug response ability (Fig. 9).
Experimental
Cell maintenance culture
Human non-small cell lung cancer cells A549 (ATCC, Manassas, VA) were cultured with DMEM/F12 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (all from Hyclone, Logan, UT). They were maintained in a humidified atmosphere of 95% air, 5% CO2 at 37 °C and their medium was refreshed every other day. All the experiments were conducted by using the cells with the passage number within 57–60.Casting of collagen gel for 3D cultivation of A549 cells
Rat tail collagen solution (BD Biosciences, San Jose, CA) at 3.4 mg ml−1 was diluted with 0.1% HAc (v/v) keeping the final concentration at 2.0 mg ml−1 for all the experiments. Before casting the 3D cultures in cell culture plate, the bottom of each well was pre-coated with the diluted collagen solution at 37 °C for 2 hours. A549 cells were then collected and resuspended within the pre-neutralized collagen solution to achieve a final concentration of 2.5 × 105 cells per ml. This concentration for seeding has been optimized in our preliminary study. The resultant cell-loaded collagen solution was then pipetted into each well for gelation at 37 °C. After 2 hours' of gelation, fresh cell culture medium was added into each well gently without disturbing the cell-loaded collagen gel. The cultures were cultivated at 37 °C in a 100% humidified incubator and the medium was refreshed every other day.DNA content assay
To determine the cellular proliferation in collagen, A549 cells were initially seeded within collagen gel at a density of ∼75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per 24 well and were cultured as described above. Cells seeded in 24 well plate without collagen gel (2D cultures) served as control. At different time points (day 1, 3, 5, 7, 9, 11), the cell-loaded collagen gels and the 2D cultures were harvested respectively and frozen at −80 °C until their DNA contents were assayed. The DNA contents were measured by PicoGreen DNA assay kit following the protocols of the manufacturer (Molecular Probes, Eugene, OR). A hybrid multi-mode microplate reader (SynergyTM H1, BioTek® Instruments, USA) was used to measure the fluorescent intensity.
000 cells per 24 well and were cultured as described above. Cells seeded in 24 well plate without collagen gel (2D cultures) served as control. At different time points (day 1, 3, 5, 7, 9, 11), the cell-loaded collagen gels and the 2D cultures were harvested respectively and frozen at −80 °C until their DNA contents were assayed. The DNA contents were measured by PicoGreen DNA assay kit following the protocols of the manufacturer (Molecular Probes, Eugene, OR). A hybrid multi-mode microplate reader (SynergyTM H1, BioTek® Instruments, USA) was used to measure the fluorescent intensity.
Calcein-AM/ethidiumhomodimer staining
For assessing cell viability by calcein-AM/ethidiumhomodimer (EthD-1) staining, cell-loaded collagen gel was first cut into 1 mm-thick samples. And then they were incubated with 0.5 ml of calcein-AM/EthD-1 staining solution at 37 °C for 2 hours as recommended by the manufacturer (Molecular Probe, Eugene, OR). After being rinsed with PBS completely, the samples were finally examined under a confocal laser scanning microscope (CLSM, Leica SP2, Heidelberger, Germany) to distinguish live cells from dead cells through their different color of fluorescence. The live cells with ubiquitous intracellular esterase activity can digest non-fluorescent calcein-AM into calcein emitting green fluorescence, whereas the dead or dying cells with damaged membranes allow the entrance of EthD-1 to bind to their nucleic acids producing a bright red fluorescence.H&E staining
Collagen-embedded A549 cells were harvested and fixed with 4% paraformaldehyde at the indicated time points. The paraffin-embedded sections were prepared by the department of pathology in the Second Medical Hospital of Dalian Medical University (Dalian, China) and stained with hematoxylin and eosin (H&E) dyes (EMD Chemicals Gibbstown, NJ). Images were captured using an Olympus upright microscope (DP72. Tokyo, Japan).RNA isolation and quantitative RT-PCR (qRT-PCR)
Total cellular RNA was extracted using RNAiso Plus reagent (Takara, Dalian, China) following the protocol provided by the manufacturer. cDNA was synthesized using a high-capacity RT-PCR kit, and real-time PCR was conducted utilized SYBR® Premix Ex Taq™ II Kit (Takara) to detect the transcript levels of EGF and EGFR, β-actin served as internal control. The primer sequences for real time PCR were listed in Table 1. The data were analyzed by Applied Biosystems StepOne™ Real-Time PCR System software version 2.0 (Applied Biosystems, Foster City, CA). The relative expression level for each target gene was normalized by the CT value of human β-actin (2ΔCT formula). Each sample was analyzed in triplicate.| Gene | Primer sequence |
|---|---|
| EGF | Forward 5′-AGCTAACCCATTATGGCAACA-3′ |
| Reverse 5′-AGTTTTCACTGAGTCAGCTCCAT-3′ | |
| EGFR | Forward 5′-AGTTTTCACTGAGTCAGCTCCAT-3′ |
| Reverse 5′-TTGGCTCACCCTCCAGAAGG-3′ | |
| β-Actin | Forward 5′-TGACAGGATGCAGAAGGAGA-3′ |
| Reverse 5′-GCTGGAAGGTGGACAGTGAG-3′ |
Western blot analysis
After 12 days of cultivation, the cultures were collected and lysed by ice-cold RIPA buffers (Sigma, USA) supplemented with proteinase inhibitor cocktail (Roche Applied Science, Mannheim, Germany) for 30 minutes. The cell lysates were harvested from the supernatant after a centrifugation at 12000 g for 30 minutes. After quantification by BCA assay kit (Beyotime, Beijing, China), a total of 20 μg protein of each sample was separated using SDS–polyacrylamide gel and transferred to a PVDF membrane (Millipore). The membrane was then incubated with anti-pho-EGFR (phosphorY1092, 1:800, Abcam, CA, UK) or anti-GAPDH (1:1000, Cell Signaling, Boston, USA), followed by incubation with the horseradish peroxidase-linked anti-rabbit secondary antibody (1:1000, Abcam). The blots were finally developed using an ECL Western-blotting protocol (Lumilight enhanced chemiluminescence, Roche). The quantification of immune reactivity was performed by densitometric scanning using the Quantity One_1-Danalysis software (Bio-Rad, Hercules, CA).
EGF content measured by ELISA
Culture medium (at day 12) was collected from the 3D cultures and the monolayer cultures (control) respectively, and was then frozen under −70 °C until assayed by ELISA. The corresponding medium was refreshed completely after each sample collection. EGF content were measured by human EGF ELISA kit (Life Tech, Rockford, IL) as recommended by the manufacturer. The amount of total protein in each group was quantitatively measured using BCA assay kit (Beyotime, Beijing, China) in order to standardize the EGF concentration measured above.Immunofluorescence staining
For immuno-fluorescence staining, the deparaffinized sections (∼5 mm) were treated with antigen retrieval solution (for antigen retrieve), 0.1% Triton-X-100 solution (for penetration) and 2% BSA solution (for blocking) sequentially (all from Fisher Scientific, Pittsburgh, PA). They were then incubated with anti-EGFR (1:50, Abcam, CA, UK) overnight at 4 °C, followed by incubation with TRITC-conjugated anti-mouse secondary antibody (1:100, Sigma) at 37 °C for 2 hours. Cell nuclei were counterstained with using 4′,6-diamidino-2-phenylindole (DAPI, Sigma). The slides were mounted in mounting media for visualization using a confocal microscope (CLSM SP8, Germany).
Scanning electron microscopy (SEM) characterization
Cultures were fixed in 2.5% glutaraldehyde in PBS overnight at 4 °C. They were then dehydrated in a series of increasing concentrations of ethanol (30%, 50%, 70%, 80%, 95% and 100%) for 15–20 minutes, respectively. After being dried in a vacuum dryer (DZF-6050, China) overnight, the samples were sputter-coated with gold particles and finally were visualized using a Hitachi S-450 SEM (Tokyo, Japan).AlamarBlue cell viability assay
Toxicity of gefitinib was evaluated through AlamarBlue cell viability assay. Firstly, gradient working solution of gefitinib (Nanjing Ange, Nanjing, China) was prepared by diluting the stock solution of gefitinib (20 mM in DMSO) with the corresponding culture medium, keeping the final concentration of DMSO vehicle below 1%. Three dimensional cultures of A549 cells in 96 well plate were then prepared as described before (80 μl cell-loaded collagen per well, 2.5 × 105 cells per ml). Monolayer cultures of A549 cells seeded in the 96 well plate (2D culture) with a same density served as control. After 48 h of cultivation with gefitinib-containing medium supplementation, AlamarBlue® assay kit (Beijing Cell Chip, Beijing, China) was used to assay the cytotoxicity of gefitinib following the manufacturer's protocol. Briefly, 10% (v/v) AlamarBlue solution was added into each well for incubation. After incubation for 4 hours, a total volume of 100 μl medium was taken out from each well and transferred into another 96 well plate for the following measurement. A hybrid multi-mode microplate reader (SynergyTM H1, BioTek® Instruments, USA) was used to measure the OD value of AlamarBlue at 570 nm with 600 nm as a reference wavelength.Growth inhibition in the 3D cultures treated by gefitinib
Effect of gefitinib on the growth of the 3D cultures was evaluated using PicoGreen DNA assay through the experimental design as followings: (1) the 3D cultures (after day 1) were treated with different concentration of gefitinib (20, 50, 100 μM diluted with culture medium) for 13 days at 37 °C in a 100% humidified incubator. Cultures without gefitinib treatment served as control. (2) The cell spheroids were first generated after 7 days of cultivation in collagen without gefitinib supplementation, then they were incubated with gefitinib solution (50, 100 μM, diluted with culture medium) for another 5 days at 37 °C in a 100% humidified incubator. The cultures without gefitinib treatment served as control. All the samples were harvested at the indicated time points and frozen at −80 °C until being assayed as described before.Statistical analysis
All experiments were carried out at least in triplicate and repeated at least two times. Unless otherwise indicated in the text, all analytical measurements were repeated three times (n = 3). All values are expressed as mean ± standard derivation (SD). Statistical analysis was performed with Student's t-test, with p < 0.05 being regarded as statistically significant.Conclusions
Taken together, an in vitro 3D culture model of NSCLCs was developed through embedding A549 cells into the collagen hydrogel. Although this bioengineered tumor models is only a simplistic representation of the complex nature of NSCLCs, several key characteristics of in vivo solid tumor, including tissue-like morphology, enhanced expression of EGF/EGFR and as well as drug-responsive capability were recapitulated. Future work is still needed to advance this in vitro bioengineered culture model into more complex, functional tumor replicates through reconstructing more pathologically relevant tumor microenvironment (incorporation more stromal cells, tumor ECM or growth factors). It is believed that this model system would be potentially utilized as a more efficient tool to evaluate a wide range of treatment modalities over conventional approaches.Acknowledgements
This work was supported by the Natural Science Foundation of Liaoning Province (2014023003), NSF of China (31470952, 81173125, 81302793, 81473181, 21572029), the National S&T Major Projects of China (2012ZX09501001, 2012ZX09506001), and the National Basic Research (2013CB531800).Notes and references
- J. S. Harunaga and K. M. Yamada, Matrix Biol., 2011, 30, 363–368 CrossRef CAS PubMed.
- S. Ishihara, H. Haga, M. Yasuda, T. Mizutani, K. Kawabata, H. Shirato and T. Nishioka, Biochem. Biophys. Res. Commun., 2010, 396, 651–655 CrossRef CAS PubMed.
- K. R. Levental, H. Yu, L. Kass, J. N. Lakins, M. Egeblad, J. T. Erler, S. F. Fong, K. Csiszar, A. Giaccia, W. Weninger, M. Yamauchi, D. L. Gasser and V. M. Weaver, Cell, 2009, 139, 891–906 CrossRef CAS PubMed.
- Y. Fu, K. Xu, X. Zheng, A. J. Giacomin, A. W. Mix and W. J. Kao, Biomaterials, 2012, 33, 48–58 CrossRef CAS PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- J. A. DiMasi and H. G. Grabowski, J. Clin. Oncol., 2007, 25, 209–216 CrossRef PubMed.
- G. Forte, S. Pagliari, M. Ebara, K. Uto, J. K. Tam, S. Romanazzo, C. Escobedo-Lucea, E. Romano, P. Di Nardo, E. Traversa and T. Aoyagi, Tissue Eng., Part A, 2012, 18, 1837–1848 CrossRef CAS PubMed.
- J. Guan, J. Yang, J. Dai, Y. Qin, Y. Wang, Y. Guo, Q. Ke and C. Zhang, RSC Adv., 2015, 5, 36175–36184 RSC.
- R. Fan, Y. Sun and J. Wan, RSC Adv., 2015, 5, 90596–90601 RSC.
- J. L. Horning, S. K. Sahoo, S. Vijayaraghavalu, S. Dimitrijevic, J. K. Vasir, T. K. Jain, A. K. Panda and V. Labhasetwar, Mol. Pharm., 2008, 5, 849–862 CrossRef CAS PubMed.
- Y. Li, Z. Zhao, J. Song, Y. Feng, Y. Wang, X. Li, Y. Liu and P. Yang, Arch. Biochem. Biophys., 2009, 490, 171–176 CrossRef CAS PubMed.
- I. Serebriiskii, R. Castello-Cros, A. Lamb, E. A. Golemis and E. Cukierman, Matrix Biol., 2008, 27, 573–585 CrossRef CAS PubMed.
- A. J. Carterson, K. Honer zu Bentrup, C. M. Ott, M. S. Clarke, D. L. Pierson, C. R. Vanderburg, K. L. Buchanan, C. A. Nickerson and M. J. Schurr, Infect. Immun., 2005, 73, 1129–1140 CrossRef CAS PubMed.
- R. C. Dutta and A. K. Dutta, Biotechnol. Adv., 2009, 27, 334–339 CrossRef CAS PubMed.
- J. Bin Kim, R. Stein and M. J. O'Hare, Breast Cancer Res. Treat., 2004, 85, 281–291 CrossRef PubMed.
- C. M. Ghajar and M. J. Bissell, Tissue Eng., Part A, 2010, 16, 2153–2156 CrossRef PubMed.
- W. Wang, G. Li, W. Zhang, J. Gao, J. Zhang, C. Li, D. Ding and D. Kong, RSC Adv., 2014, 4, 30168–30171 RSC.
- N. Li, D. Wang, Z. Sui, X. Qi, L. Ji, X. Wang and L. Yang, Tissue Eng., Part C, 2013, 19, 708–719 CrossRef CAS PubMed.
- X. Wang, L. Sun, M. V. Maffini, A. Soto, C. Sonnenschein and D. L. Kaplan, Biomaterials, 2010, 31, 3920–3929 CrossRef CAS PubMed.
- U. Fernekorn, J. Hampl, C. Augspurger, C. Hildmann, F. Weise, M. Klett, A. Laeffert, M. Gebinoga, A. Williamson and A. Schober, RSC Adv., 2013, 3, 16558–16568 RSC.
- K. Ruan, G. Song and G. Ouyang, Journal of cellular biochemistry, 2009, 107, 1053–1062 CrossRef CAS PubMed.
- S. B. Leite, A. P. Teixeira, J. P. Miranda, R. M. Tostoes, J. J. Clemente, M. F. Sousa, M. J. Carrondo and P. M. Alves, Toxicology, 2011, 25, 825–832 CAS.
- X. L. Zhang, W. Wang, W. T. Yu, Y. B. Xie, X. H. Zhang, Y. Zhang and X. J. Ma, Biotechnol. Prog., 2005, 21, 1289–1296 CrossRef CAS PubMed.
- R. Ng, R. Zang, K. K. Yang, N. Liu and S.-T. Yang, RSC Adv., 2012, 2, 10110–10124 RSC.
- D. E. Ingber, Semin. Cancer Biol., 2008, 18, 356–364 CrossRef CAS PubMed.
- G. Chen, J. Chen, Q. Liu, C. Ou and J. Gao, RSC Adv., 2015, 5, 30675–30678 RSC.
- E. E. Antoine, P. P. Vlachos and M. N. Rylander, Tissue Eng., Part B, 2014, 20, 683–696 CrossRef CAS PubMed.
- J. H. Lee, J.-H. Park, M. Eltohamy, R. Perez, E.-J. Lee and H.-W. Kim, RSC Adv., 2013, 3, 24202–24214 RSC.
- A. K. Larsen, D. Ouaret, K. El Ouadrani and A. Petitprez, Pharmacol. Ther., 2011, 131, 80–90 CrossRef CAS PubMed.
- N. Normanno, C. Bianco, A. De Luca and D. S. Salomon, Front. Biosci., 2001, 6, D685–D707 CrossRef CAS.
- J. A. Kiefer and M. C. Farach-Carson, Matrix Biol., 2001, 20, 429–437 CrossRef CAS PubMed.
- M. J. Paszek, N. Zahir, K. R. Johnson, J. N. Lakins, G. I. Rozenberg, A. Gefen, C. A. Reinhart-King, S. S. Margulies, M. Dembo, D. Boettiger, D. A. Hammer and V. M. Weaver, Cancer Cell, 2005, 8, 241–254 CrossRef CAS PubMed.
- L. David, V. Dulong, D. Le Cerf, L. Cazin, M. Lamacz and J. P. Vannier, Acta Biomater., 2008, 4, 256–263 CrossRef CAS PubMed.
- Z. F. Cui, X. Xu, N. Trainor, J. T. Triffitt, J. P. Urban and U. K. Tirlapur, Toxicology, 2007, 21, 1318–1324 CAS.
- Y. J. Kim, H. I. Bae, O. K. Kwon and M. S. Choi, Int. J. Biol. Macromol., 2009, 45, 65–71 CrossRef CAS PubMed.
- F. M. Balis, J. Natl. Cancer Inst., 2002, 94, 78–79 CrossRef PubMed.
- K. Bhadriraju and C. S. Chen, Drug Discovery Today, 2002, 7, 612–620 CrossRef CAS PubMed.
- O. Tredan, C. M. Galmarini, K. Patel and I. F. Tannock, J. Natl. Cancer Inst., 2007, 99, 1441–1454 CrossRef CAS PubMed.
Footnote |
| † D.-D. Wang and W. Liu sharing equally contribution to this work. |
| This journal is © The Royal Society of Chemistry 2016 |