
Vibrational spectroscopic and bond valence study of structure and bonding in Al2O3-containing AgI–AgPO3 glasses
Abstract
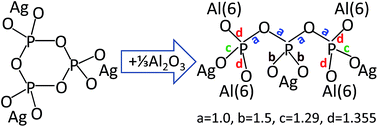
We present a detailed investigation of the effects of synthesis conditions on glasses xAgI–(1 − x)AgPO3 with 0 ≤ x ≤ 0.4. Raman and infrared spectroscopy of glasses synthesized in platinum crucibles showed that their metaphosphate structure remains largely unaffected by increasing the amount of AgI. This result contradicts recent findings on similar glasses synthesized by melting in alumina crucibles, which were found to exhibit a strongly changing phosphate structure with addition of AgI. The glass transition temperature and ionic conduction properties of the latter glasses also show strong deviation from the analogous properties of glasses melted in platinum crucibles, as in this work. To reveal the origin of such effects, glasses with the nominal composition 0.4AgI–0.6AgPO3 were synthesized in alumina crucibles and their structures were compared with glasses prepared in platinum crucibles with composition yAl2O3–(1 − y)[0.4AgI–0.6AgPO3] and y = 0.0, 0.04 and 0.07. Vibrational spectroscopy showed that melting in alumina crucibles, especially in the presence of P2O5, leads to doping the AgI–AgPO3 glass with Al2O3 leached from the crucible. The bond valence approach was employed to assist assigning the new Raman bands that develop when Al2O3 modifies the metaphosphate structure, with an exponential relationship describing the dependence of the P–O stretching frequency on bond strength. Combination of this relationship with bond valence principles allowed us to associate the formation of aluminum-triphosphate species Al2/3Ag3P3O10 with the Raman trends observed for AgI–AgPO3 glasses doped with Al2O3 either intentionally or unintentionally, i.e. when melted in alumina crucibles for long times.
 Please wait while we load your content...
Please wait while we load your content...

