
Density functional theory study of Li binding to graphene†
Abstract
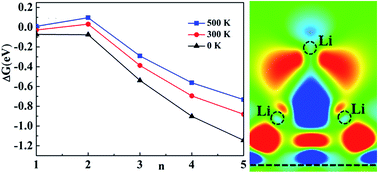
Using first-principle calculations, we studied the interaction between Li and graphene by considering two kinds of models, which are related to the configurations of Li adsorption and the concentration of Li on graphene. In a low concentration, the 2s state of Li is fully unoccupied due to charge transfer. With the increase of Li concentration, the 2s state is broadened and occupied partly by electrons. With a high concentration, such as Li : C = 1 : 6, Li cluster adsorption seems to become popular by the free formation energy of clusters with thermal effects.
 Please wait while we load your content...
Please wait while we load your content...

