
Novel thiosemicarbazide–oxadiazole hybrids as unprecedented inhibitors of yeast α-glucosidase and in silico binding analysis†
Muhammad Taha*ab,
Nor Hadiani Ismailab,
Syahrul Imranab,
Abdul Wadoodc,
Muhammad Alid,
Fazal Rahime,
Aftab Ahmad Khane and
Muhammad Riazc
aAtta-ur-Rahman Institute for Natural Product Discovery, Universiti Teknologi MARA (UiTM), PuncakAlam Campus, 42300 Bandar PuncakAlam, Selangor, Malaysia. E-mail: taha_hej@yahoo.com; muhamm9000@puncakalam.uitm.edu.my; Tel: +60 183098141
bFaculty of Applied Science UiTM, 40450 Shah Alam, Selangor, Malaysia
cDepartment of Biochemistry, Computational Médicinal Chemistry Laboratory, Abdul Wali Khan University, Mardan, Khyber Pukhtunkhwa, Pakistan
dDepartment of Chemistry, COMSATS Institute of Information Technology, University Road, Abbottabad-22060, KPK, Pakistan
eDepartment of Chemistry, Hazara University, Mansehra, Khyber Pukhtunkhwa, Pakistan
First published on 22nd March 2016
Abstract
Compounds 1–18, new oxadiazole–thiosemicarbazide hybrids, were synthesized using a five-step reaction sequence in excellent yields. All the synthesized analogs exhibited exceptional α-glucosidase inhibitory potentials in the range of 0.4–38.1 μM. Among the current series, it was observed that the fluoro-substituted analogues were exceptionally potent inhibitors of α-glucosidase. The study provides a proof of concept that inductively strong electron withdrawing groups result in enhanced inhibitory potentials. The binding interactions of these compounds were analyzed in silico for possible prediction and identification of the improved inhibitory potential.
1. Introduction
The α-glucosidase inhibitors have attracted the attention of medicinal chemists because of their key biological role in glycoprotein processing and carbohydrates digestion.1,2 α-Glucosidase is membrane-bound enzyme located at the epithelium of the small intestine.3 α-Glucosidase is responsible for catalyzing the final step in the digestive process of carbohydrates.4–6 In addition, α-glucosidase inhibitors can decrease the gastrointestinal absorption of dietary carbohydrates by inhibiting the digestion of polysaccharides and disaccharides and thus reducing the postprandial increase in blood glucose levels.6,7 Several sugar α-glucosidase inhibitors, such as acarbose, voglibose, and miglitol, are clinically used in the effective treatment of type-2 diabetes mellitus.8Diabetes mellitus is a metabolic disorder of multiple etiologies characterized by chronic hyperglycemia with disturbances of carbohydrate, fat and protein metabolism resulting from defects in insulin secretion (Type-1 diabetes), insulin action (Type-2 diabetes) or both. As a result, elevated blood sugar levels lead to the common effects of uncontrolled diabetes with serious damage to many of the body's systems, especially the nerves and blood vessels. Type-2 diabetes mellitus accounts for approximately 80–90% of all cases of diabetes and it is the fastest growing global threat to public health.9 Therefore, effective therapies for diabetes and its complications require the simultaneous suppression of hyperglycemia.10
Thiosemicarbazides have found a significant place in drug discovery and industry. Furthermore, this class of compounds has been utilized in the synthesis of biologically active compounds, viz., triazoles and thiazoles. It has been revealed that the biological potentials of these compounds are associated with their oxidation mechanism. It is also generally accepted that the criterion for thio-compounds to express their physiological effects is through S-oxidation11 because the oxidation of organosulfur compounds appears to be involved in many cellular functions,12 including the reductive degradation of polypeptide hormones and proteins, regulation of protein synthesis, maintenance of intracellular redox potential, and protection of cell from oxidative damage. Thiosemicarbazide and its derivatives are of colossal curiosity pertaining to their wide synthetic and analytical applications along with their biological activities.13 Thiosemicarbazides derivatives display interesting biological profiles, including anticancer,14 anti HIV,15 antibacterial,16 antiviral17 and antifungal properties owing to their ability to diffuse through the semipermeable membrane of cell lines.18 They play a pivotal role in the regulation of plant growth.19 The ease of synthesis combined with their abundance in plants has made this class of compounds potential therapeutic agents. Because thiosemicarbazides are also sulfur and nitrogen donor ligands, their co-ordination complexes have attained considerable attention owing to their biological potentials against protozoa, influenza, small poxvirus, fungi and cancer. Some industrially important activities, such as anticorrosion and antifouling effects,20 have also been observed for these compounds. Furthermore, thiosemicarbazides are also known as antidotal against metal toxicity.21
A lot of attention has been paid to the chemistry and biological activities of 1,3,4-oxadiazole moieties. Several compounds possessing 1,3,4-oxadiazole scaffolds have been recently reported as potential antiproliferative agents.22–24 Apart from anticancer activity, other biological activities have also been reported for 1,3,4-oxadiazole derivatives such as antidiabetic,25 antitubercular,26 β-glucuronidase inhibitory potential,27 antioxidant,28 antifungal,29 anti-inflammatory,30 and antibacterial activities.31
Thiosemicarbazides and oxadiazoles, individually, are useful structural motifs and have great biological potential. Optimization of their structural hybrids may lead to groundbreaking discoveries of new classes of therapeutic representatives. As a part of our study in the pursuit of novel lead molecules, we herein report the synthesis of hybrid-scaffolds containing oxadiazole and thiosemicarbazide moieties, their characterization, evaluation of α-glucosidase inhibitory potentials, in silico investigations and cytotoxicity analysis.
2. Results and discussion
2.1 Chemistry
The synthetic approaches used to prepare the oxadiazole–thiosemicarbazide hybrids 1–18 are shown in Schemes 1 and 2. Methyl 2-chlorobenzoate (I) was refluxed with methanolic hydrazine hydrate solution to obtain hydrazide (II). The hydrazide (II) was then condensed with methyl 4-formylbenzoate in methanol, while using a catalytic amount of acetic acid to afford hydrazone (III), which was then subjected to an oxidative cyclization using phenyliododiacetate [PhI(OAc)2] in dry chloroform to afford oxadiazole (IV). The ester functional group of oxadiazole (IV) was further transformed into hydrazide upon treatment with hydrazine hydrate to provide the oxadiazole–hydrazide hybrid (V).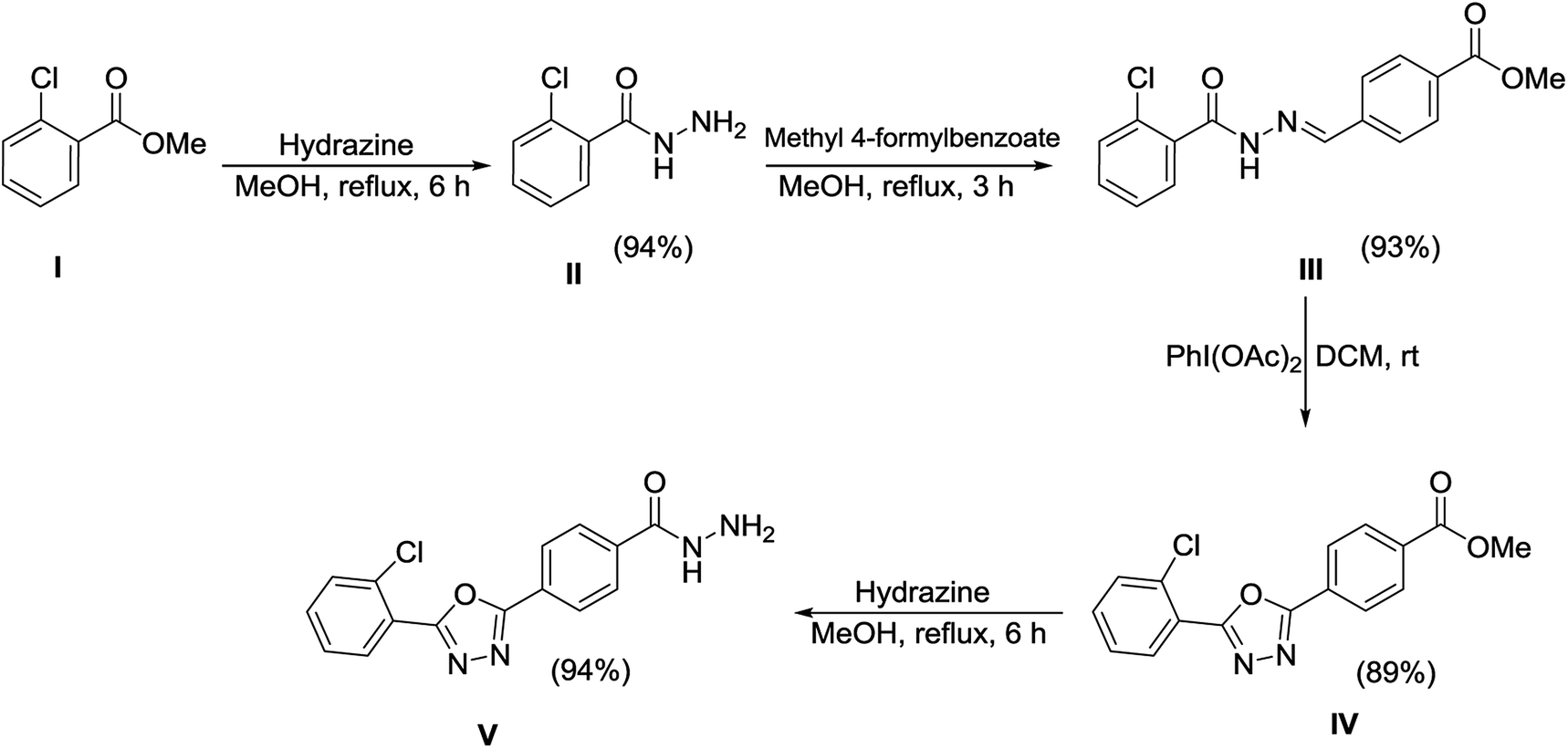 | ||
| Scheme 1 Synthesis of oxadiazole–hydrazide hybrid (V) via a four-step reaction sequence. | ||
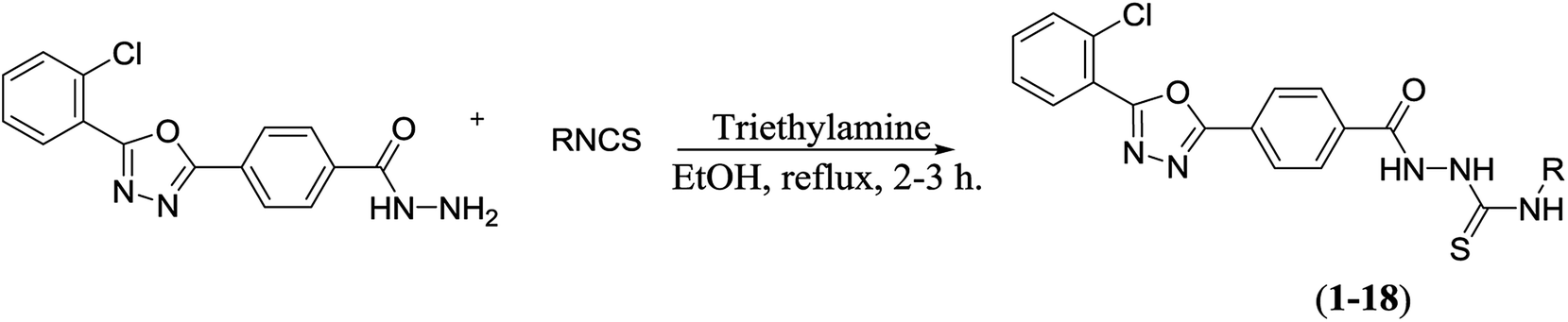 | ||
| Scheme 2 Synthesis of the oxadiazole–thiosemicarbazide hybrids 1–18. | ||
The oxadiazole–hydrazide hybrid (V) was further reacted with various isothiocyanates in the presence of a catalytic amount of triethylamine (TEA) to yield the oxadiazole–thiosemicarbazide hybrids 1–18 (Table 1 and Scheme 2). The structures of the synthesized compounds 1–18 were confirmed using spectroscopic techniques, such as NMR, and MS, and were further confirmed using elemental analysis.
| S. No | R | Yield (%) | S. No | R | Yield (%) | S. No | R | Yield (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | 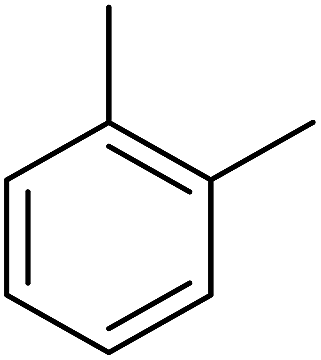 |
84 | 7 | 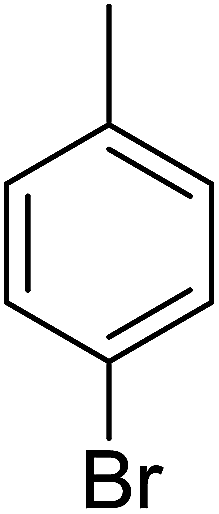 |
85 | 13 | 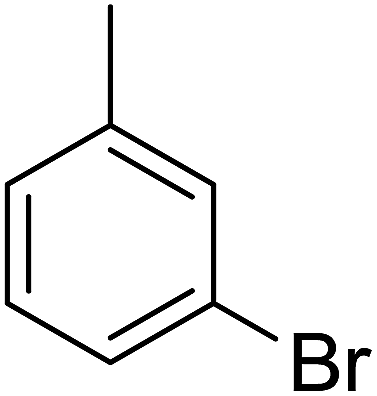 |
85 |
| 2 | 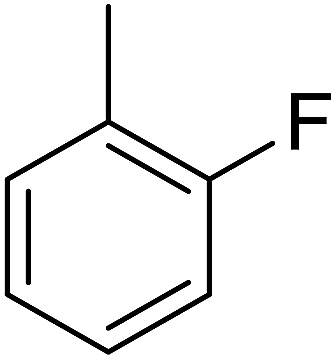 |
88 | 8 | 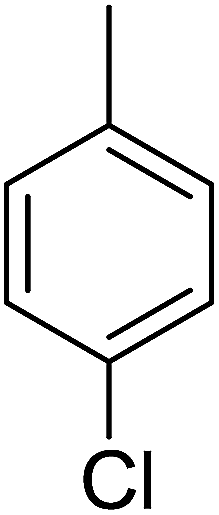 |
87 | 14 | 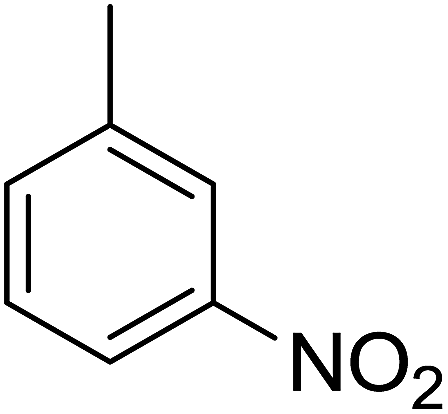 |
85 |
| 3 | 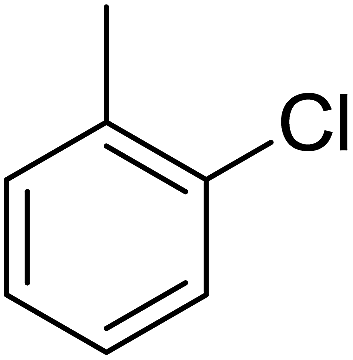 |
86 | 9 | 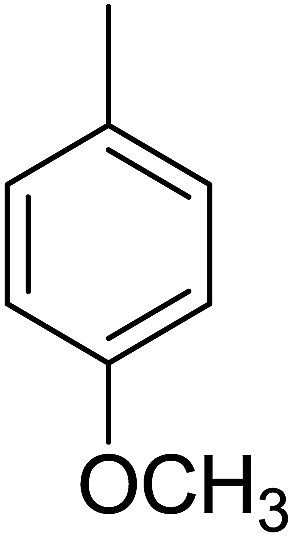 |
87 | 15 | 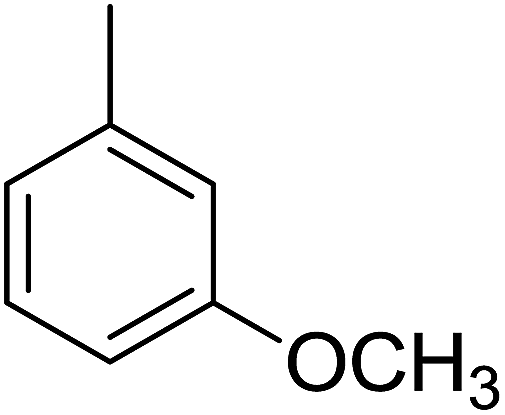 |
86 |
| 4 | 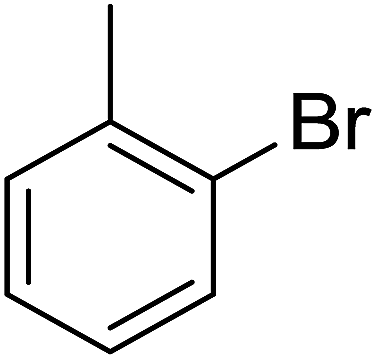 |
85 | 10 | 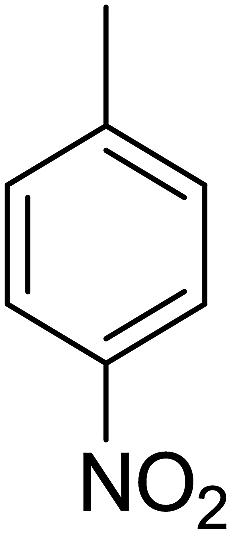 |
89 | 16 | 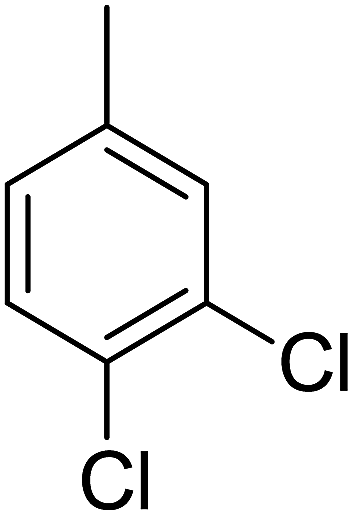 |
86 |
| 5 | 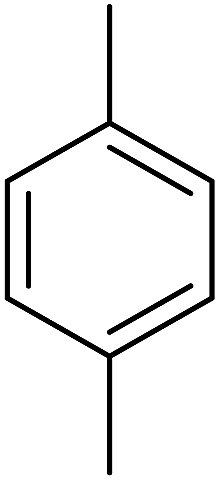 |
87 | 11 | 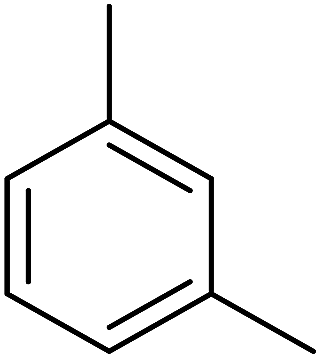 |
84 | 17 | 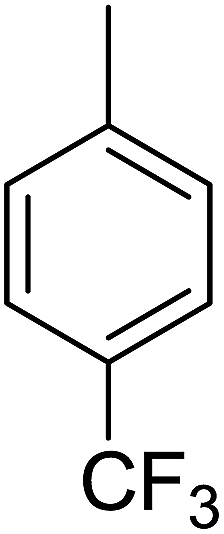 |
82 |
| 6 | 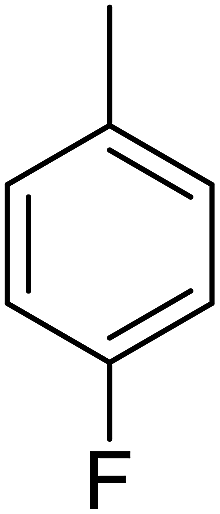 |
86 | 12 | 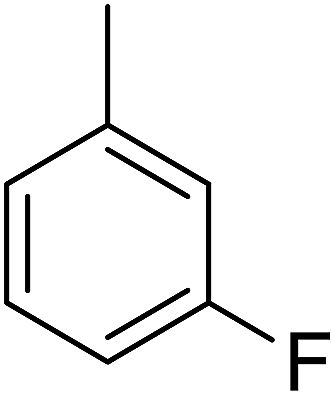 |
83 | 18 | 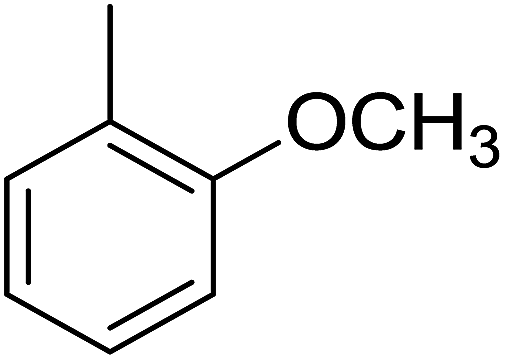 |
84 |
2.2 Biological evaluation
The concept of hybrid molecules has gained importance over the last decade, whereas pharmacophoric hybridization is emerging as an alternative to the existing strategies used for drug design and development. These medicinal hybrids, also known as “dual drugs or double drugs”, are particularly effective in overcoming the issues related to drug resistance. Furthermore, an enhancement of biological potential (synergism) is another advantage of the hybrid molecular structure approach.32 This manuscript unfolds the α-glucosidase inhibitory potentials of oxadiazole–thiosemicarbazide hybrids 1–18 in search of new and potential leads.Compounds 1–18 were screened for yeast α-glucosidase inhibitory potential according to literature protocol.33 All the synthetic analogs exhibited excellent inhibitory potentials against the α-glucosidase enzyme. The inhibitory potentials were found in the range of 0.4–38.1 μM when compared with standard inhibitor acarbose (IC50 value 38.25 ± 0.12 μM) (Table 2). The most active inhibitor 2 was found to be ∼95 times more active than the standard inhibitor, whereas the least active, 15, was found equivalent in inhibitory potential to that of the standard inhibitor acarbose.
| S. No. | IC50 (μM ± SEMa) | S score | S.No. | IC50 (μM ± SEMa) | S score |
|---|---|---|---|---|---|
| 1 | 1.4 ± 0.01 | −11.5481 | 10 | 10.4 ± 0.10 | −11.1203 |
| 2 | 0.4 ± 0.001 | −12.2285 | 11 | 18.7 ± 0.14 | −10.8321 |
| 3 | 0.7 ± 0.002 | −11.8805 | 12 | 3.3 ± 0.02 | −11.5853 |
| 4 | 4.9 ± 0.02 | −11.1343 | 13 | 31.4 ± 0.32 | −10.3019 |
| 5 | 10.4 ± 0.18 | −11.0035 | 14 | 15.7 ± 0.27 | −11.4100 |
| 6 | 2.5 ± 0.02 | −11.4349 | 15 | 38.1 ± 0.38 | −10.0021 |
| 7 | 23.7 ± 0.25 | −10.8682 | 16 | 1.1 ± 0.01 | −11.8130 |
| 8 | 5.5 ± 0.06 | −11.1280 | 17 | 0.6 ± 0.001 | −12.2118 |
| 9 | 33.6 ± 0.36 | −10.5448 | 18 | 5.6 ± 0.08 | −11.1163 |
| Acarbose | 38.25 ± 0.12 μM | ||||
Structure–activity relationships suggested that the activity of a specific molecule was seemingly directed by the substitution present on the aromatic residue attached with the terminal nitrogen of thiosemicarbazide part of the hybrid structure. The ortho-methyl substituted analog 1 (IC50 = 1.4 ± 0.01 μM), ortho-fluoro analog 2 (IC50 = 0.4 ± 0.001 μM), ortho-chloro analog 3 (IC50 = 0.7 ± 0.002 μM), ortho-bromo analog 4 (IC50 = 4.9 ± 0.02 μM), para-fluoro analog 6 (IC50 = 2.5 ± 0.02 μM), para-chloro analog 8 (IC50 = 5.5 ± 0.06 μM), meta-fluoro analog 12 (IC50 = 3.3 ± 0.02 μM), 3,4-dichloro analog 16 (IC50 = 1.1 ± 0.01 μM), para-trifluoromethane analog 17 (IC50 = 0.60 ± 0.001 μM) and ortho-methoxy analog 18 (IC50 = 5.60 ± 0.08 μM) showed remarkable inhibitory potentials among the series. It was observed that variation in the substitution pattern of the aromatic regions are responsible for the inhibitory potential of the scaffolds. Compound 2 (ortho-fluoro analog) was found to be the most potent analog among the series with an IC50 value of 0.4 ± 0.001 μM (∼95 times more active than the standard). The potent activity of this molecule may be attributed to the fluoro group. Compound 17 was the second most active among the series with an IC50 value of 0.60 ± 0.001 μM. This compound possesses a trifluoromethane group in the para position of the aromatic ring, which might be responsible for good inhibition. Similarly, compounds 6 and 12 with para-fluoro and meta-fluoro substitutions have IC50 values of 2.5 ± 0.02 and 3.3 ± 0.02 μM, respectively, which showed very potent inhibition properties. These observations suggest the strong electron withdrawing nature of the fluorine atom played a pivotal role in the inhibition potential of the fluoro substituted analogs. The presence of strong electron withdrawing groups such as fluorine, as in our case, polarized the molecules in a way that made the hydrogens present on the thiosemicarbazide part more polar or acidic. These acidic hydrogens have the potential to form stronger non-bonded interactions with their nearby receptor's counterparts, which results in the higher inhibitory potentials. Compound 3, an ortho-chloro analog, was the third most active analog among the series. A comparison between compound 3, compound 8 (IC50 value 5.5 ± 0.06 μM) and 16 (IC50 value 1.1 ± 0.01 μM) showed that compound 3 was at least two times more active than 8 and 16. This pattern of inhibitory potential for the fluoro- and chloro-derivatives seems to be very interesting because the electronegativity of both atoms (F and Cl) and their corresponding electron withdrawing inductive effects played a substantive role. In a nut shell, the greater the polarization of the molecule, the more active it will be.
Compound 1, an ortho-methyl analog with an IC50 value of 1.4 ± 0.01 μM, 5, a para-methyl analog with an IC50 value of 10.4 ± 0.18 μM and 11, a meta-methyl analog with an IC50 value of 18.7 ± 0.14 μM. The great activity difference between these compounds was mainly due to the position of the substituents. Compound 9, a para-methoxy analog with an IC50 value 33.6 ± 0.36 μM, 15, a meta-methoxy analog with an IC50 value of 38.12 ± 0.38 μM and 18, an ortho-methoxy analog with an IC50 value of 5.60 ± 0.08 μM. It can be observed that both an electron withdrawing group and electron donating group in the ortho-position showed great inhibition but the EWG are superior to the EDG. In the case of the bromo-substituted analogs 4, 7 and 13 and nitro-substituted analogs 10 and 14, we have observed similar results. The binding interactions of the most active compounds were confirmed through molecular docking studies.
2.3 Molecular docking experiments
The Molecular Operating Environment (MOE) docking program [Molecular operating environment MOE. 2008. C.C.G.I.M, Quebec Canada, MOE-Dock, Chemical Computing Group. 1998. Inc., Montreal Quebec Canada.] was utilized to analyze the binding modes of the eighteen hybrid molecules, synthesized from thiourea and oxadiazole, against the α-glucosidase enzyme. To obtain the minimum energy structures, the ligands were allowed to be flexible during docking. The ligands are ranked using the scores obtained from the GBVI/WSA binding free energy calculation in the S field, i.e., the score of the last stage. The GBVI/WSA is a scoring function, which estimates the free energy of binding of the ligand from a given pose. For all scoring functions, lower scores indicate more favorable poses. The unit for all the scoring functions is kcal mol−1.34 The default parameters of the MOE-Dock program were used for the molecular docking of the synthesized compounds. The top ranked conformation of each compound was selected on the basis of the docking score (S) for further analysis. At the end of docking, the best conformations on the basis of the docking score were analyzed for hydrogen bonding/arene–arene and arene–cation interactions. Analysis of the predicted binding conformations showed that the compounds in which the substituent has either EWG or EDG in the ortho position showed good in silico inhibition, but the EWG are superior to EDG. For example, the most active compound 2 with a fluorine (F) group in the ortho position (IC50 = 0.4 ± 0.001) showed the best interactions (Fig. 1a) among the series of the eighteen synthesized compounds. His279 and Arg439 were shown to be involved in the arene–cation interactions with the chlorobenzene and fluorobenzene moieties, respectively, and His245 showed hydrogen bonding with the oxadiazole moiety. In addition, the central phenyl ring of the compound was found to make arene–arene interactions with the hydrophobic residue Phe157 of the α-glucosidase enzyme.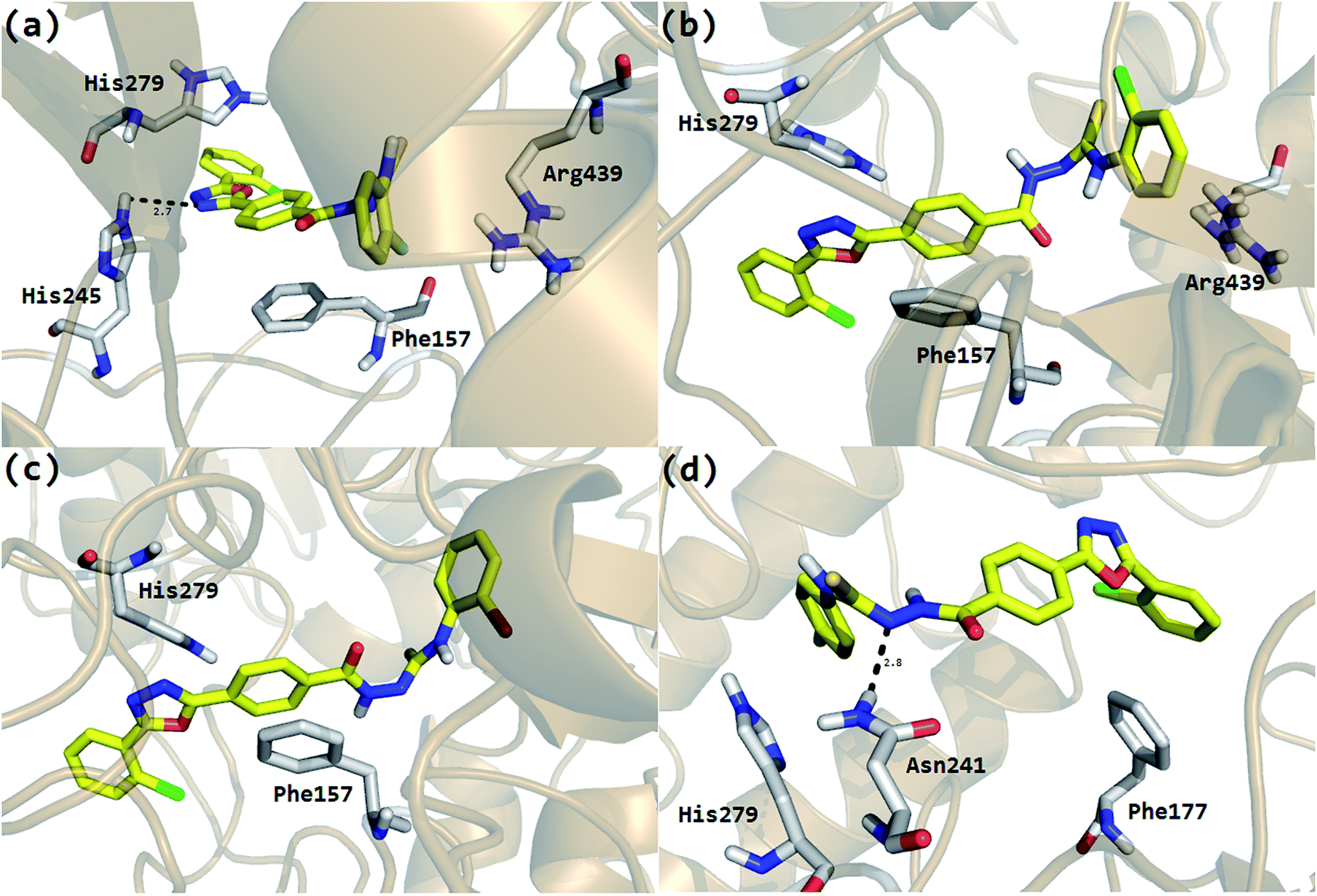 | ||
| Fig. 1 The molecular interactions and surface visualization of the hybrid molecules synthesized from thiourea and oxadiazole in the active site of the α-glucosidase enzyme (predicted model). (a) Compound 2, (b) compound 3, (c) compound 4 and (d) compound 6. The ligand is shown as a yellow stick (molecular color). | ||
In case of compound 3 and 4, the halogens, chlorine and bromine, occupy the ortho position in the phenyl ring and also showed good interactions, but slightly inferior to compound 2. In the case of compound 3, Phe157 made arene–arene contacts with the central phenyl ring, His279 interacts with the oxadiazole moiety and Arg439 with chlorobenzene (Fig. 1b). Exactly similar contacts with His279 and Phe157 were observed for compound 4 (Fig. 1c). The docking study predicted almost the same behavior as that observed in the biological evaluations of the different halogen groups (F, Cl, Br) in the ortho position of the substituted phenyl group. In the biological evaluations, the ortho-fluoro analog 2 (IC50 = 0.4 ± 0.001 μM) was found to be superior to the ortho-chloro analog 3 (IC50 = 0.7 ± 0.002 μM) and ortho-bromo analog 4 (IC50 = 4.9 ± 0.02 μM) and the same order was found in these analogs regarding interactions and docking scores (Table 2).
It was also observed that as the position of the substituent in the phenyl ring goes on changing, i.e., from ortho to para and meta, their biological activities as well as their interaction patterns and docking scores also change. For example, compound 6 that has fluorine in the para-position of the phenyl ring showed good mode of interaction, but was lower when compared to compound 2 that has fluorine in the ortho-position. Phe177 showed an arene–arene contact with chlorobenzene, His279 with fluorobenzene and Asn 241 showed hydrogen bonding with the N of the urea moiety (Fig. 1d). Similarly, compound 7 with bromine in the para position showed a slightly lower interaction pattern (Phe177 and His279) when compared to compound 4 (ortho-bromo analog) and almost similar behavior was found between compound 8 with chlorine in the para position (Phe157) and compound 3.
Similarly, a comparatively lower inhibition potential for the meta substituted moieties 12, 13, 14 and 15 was observed computationally when compared to the ortho and para substituted moieties, which was in accordance with their biological activities determined experimentally [Table 2].
From the structural differences of these compounds and their molecular docking studies, it was observed that the compounds with an ortho substituent in the aromatic ring have a good interaction network, as well as inhibitory activities when compared to the compounds with either meta or para substituents. Similarly, a good in silico inhibition mode was observed for most electronegative substituents compared to the less electronegative substituents on the aromatic phenyl ring.
3. Conclusions
In a nut shell, compounds 1–18, synthesized using a five-step reaction sequence demonstrated a great deal of α-glucosidase inhibitory potential. Compounds 1, 2, 3, 4, 6, 8, 12, 16, 17 and 18 were found to be remarkably potent inhibitors of α-glucosidase with IC50 values of 1.4 ± 0.01, 0.4 ± 0.001, 0.7 ± 0.002, 4.9 ± 0.02, 2.5 ± 0.02, 5.5 ± 0.06, 3.3 ± 0.02, 1.1 ± 0.01, 0.60 ± 0.001 and 5.60 ± 0.08 μM, respectively, when compared with the standard inhibitor (acarbose: IC50 value 38.25 ± 0.12 μM). The structure–activity relationships showed that the presence of a strong electron withdrawing group, such as fluoro and chloro groups, resulted in enhanced activity. The binding interactions of the compounds with their receptors were predicted via molecular docking. All the synthesized compounds 1–18 were tested for cytotoxicity but none of them were found to be toxic.4. Experimental
4.1 General procedures
Melting points were measured in open glass capillaries using a Stuart Scientific SMP11 Analog melting point apparatus. NMR spectroscopy of the synthesized derivatives 1–18 was performed on a Bruker Ultra Shield FT NMR 500 MHz spectrometer. EI-MS was carried out using a Finnigan-MAT-311 A instrument. Thin layer chromatography was carried out on precoated silica gel plates (Merck, Kieselgel 60 F-254, 0.20 mm) and visualized using a UV lamp at 254 nm (UVGL-58; Upland, USA). Elemental analysis were performed on a Carlo Erba Strumentazion-Mod-1106 instrument.4.2 α-Glucosidase inhibitory assay
The α-glucosidase inhibition activity assay was performed according to a literature procedure with slight modifications.31 A total volume of 100 μL reaction mixture contained 70 μL of 50 mM phosphate buffer at pH 6.8, 10 μL (0.5 mM in methanol) of test compound, followed by the addition of 10 μL (0.057 units, Sigma Inc.) of enzyme solution in the buffer. The contents were mixed, pre-incubated for 10 min at 37 °C and pre-read at 400 nm. The reaction was initiated by the addition of 10 μL of 0.5 mM substrate (p-nitrophenylglucopyranoside, Sigma Inc.). After 30 min of incubation at 37 °C, the absorbance of p-nitrophenol was measured at 400 nm using the Synergy HT 96-well plate reader, BioTek, USA. Acarbose was used as a positive control. All experiments were carried out in triplicate (mean ± SEM, n = 3). The percentage inhibition was calculated using the following equation:| Inhibition (%) = (abs of control − abs of test/abs of control) × 100 |
Active compound solutions were suitably diluted and their inhibition studies were determined. The data obtained was used for determination of the IC50 values (concentration at which there is 50% enzyme inhibition) using EZ-Fit Enzyme Kinetics Software (Perrella Scientific Inc. Amherst, USA).
4.3 Cytotoxicity assays using 3T3-L1 and CC-1 cell-lines and MTT
In vitro cytotoxicity assays were performed as described by Taha et al., (2015),35 using the 3T3-L1 mouse embryo fibroblast cell line (American Type Culture Collection ‘ATCC’, Manassas, VA 20108, USA) and CC-1 cells, a rat Wistar hepatocyte cell line (European Collection of Cell Cultures, Salisbury, UK). The CC-1 cells were suspended in Minimum Essential Medium Eagle (MEM) supplemented with 10% FBS, 2 mM glutamine, 1% non-essential amino acids and 20 mM HEPES. While the 3T3-L1 cells were suspended in Dulbecco's Modified Eagle's Medium (DMEM) formulated with 10% FBS. Using flat bottomed plates, both cell-lines were plated at a concentration of 6 × 104 cells per mL and incubated for 24 h at 37 °C under a 5% CO2 environment. After removal of the media, the cells were challenged with three different concentrations (1.0, 5.0, and 20 μg mL−1) of the compounds in triplicate and were then further incubated for 48 h at 37 °C in a CO2 incubator. Following exposure to each compound, the cells viability was assessed using 0.5 mg mL−1 of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) for 4 h followed by removal of supernatant and the addition of DMSO to solubilize the formazan complex. Plates were read at 540 nm after one minute of shaking and the readings were processed using MS Excel software. The results were expressed as the mean ± standard deviation for the triplicate readings.4.4 Molecular docking calculations
The study was designed to dock hybrid molecules obtained from thiourea and oxadiazole against the α-glucosidase enzyme with the following communications; Intel® Xenon® CPU E5620@2.40 GHz system having 3.8 GB RAM with the open 11.4 (X 86_64) operating platform. Protein-Ligand docking was carried out using the Molecular Operating Environment (MOE 2010.11) software package. The three dimensional structure of α-glucosidase from Saccharomyces cerevisiae has not been solved up-to yet. Although only few homology models have been reported,36–39 in the current study we predicted the 3D structure of α-glucosidase from Saccharomyces cerevisiae using the same protocol as described in the literature38 for homology modeling. The primary sequence of α-glucosidase for Saccharomyces cerevisiae was retrieved from UniProt (access code P53341) and a template search was performed using Protein BLAST against the PDB. The crystallographic structure of Saccharomyces cerevisiae isomaltase (PDB code 3AJ7; resolution 1.30 Å) with 72.4% of sequence identity with the target was selected as a template due to its highest sequence identity among the given templates.40 The 3D structure of α-glucosidase from Saccharomyces cerevisiae was predicted using MOE homology modeling tools. A set of 10 intermediate models were generated and refined with AMBER99 force field and the best of them was selected and evaluated. The developed model was then subjected to energy minimization up to 0.05 gradients and the quality of the modeled structure was assessed by Ramachandran and ProSA plots. The evaluation of the backbone Psi and Phi dihedral angles for the α-glucosidase model revealed that 94.8% of the residues lie in the favored region, 4.8% of the residues lie in the allowed region and only 0.3% of the residues lie in the outlier region. Analysis of ProSA shows a Z-value of −10.76, indicating no significant deviation from the score determined for a protein of similar size. The results of both the Ramachandran and ProSA plots reflect the accuracy of our modeled structure used in the docking protocol.Before docking, the ligands and protein were prepared using MOE v2010.11. The 3D structures of all the compounds were built using the Molecular Builder Module program implemented in MOE and were saved as a (.mdb) file for molecular docking. Subsequently, the energy of all the compounds was minimized up to 0.05 Gradient using MMFF94 s force field.41 Energy minimization of the compounds was followed by the preparation of the protein for docking purposes. Most macromolecular crystal structures contain little or no hydrogen coordinate data due to limited resolution and thus protonation was carried out prior to docking using Protonate 3D tools. Protonation was followed by energy minimization up to 0.05 Gradient using Amber 99 force field.42 All the compounds were docked into the active site of the protein using the Triangular Matching docking method (default), which generate poses by aligning ligand triplets of atoms on triplets of alpha spheres in a more systematic way than in the Alpha Triangle method, and 30 conformations of all the compounds and protein complex were generated with a docking score (S). The complex was analyzed for interactions and their 3D images were taken using the visualizing tool PyMol (The Pymol Molecular Graphics System, version 1.7.4 Schrodinger, LLC).
4.5 Synthesis of 2-chlorobenzohydrazide (II)
Methyl 2-chlorobenzoate (1) (30 mmol) and 30 mL of hydrazine hydrate were mixed in methanol (30 mL). The mixture was refluxed for 4 hours. Methanol was then evaporated and the product formed was washed with plenty of water to remove the excess hydrazine hydrate. The product formed was left to dry at room temperature and was obtained in a 94% yield. mp: 240–241 °C, 1H NMR (500 MHz, DMSO-d6): δ 9.58 (s, 1H, NH), 7.50 (d, J = 8.0 Hz, 1H), 7.46–7.42 (m, 1H), 7.39 (dd, J = 8.0, 2.0 Hz, 1H), 7.36 (t, J = 7.0 Hz, 1H), 4.50 (s, 2H, NH2), 13C NMR (150 MHz, DMSO-d6): δ 163.1, 135.2, 132.1, 131.2, 130.7, 126.6, 126.0, HR-ESI-MS: m/z calcd for C7H7ClN2O, [M]+ 170.0247; found 170.0251, anal. calcd for C7H7ClN2O, C = 49.28; H = 4.14; N = 16.42; found C = 49.29; H = 4.12; N = 16.43.Acknowledgements
The authors would like to acknowledge the Universiti Teknologi MARA for financial support under the Research Intensive Faculty grant scheme, reference number UiTM 600-RMI/DANA 5/3/LESTARI (54/2015).References
- E. Gallienne, T. Gefflaut and M. Lemaire, J. Org. Chem., 2006, 71, 894 CrossRef CAS PubMed.
- J. E. Groopman, Rev. Infect. Dis., 1990, 12, 908 CrossRef CAS PubMed.
- A. J. Hirsh, S. Y. Yao, J. D. Young and C. I. Cheeseman, Gastroenterology, 1997, 113, 205 CrossRef CAS.
- H. Gao, Y. N. Huang, P. Y. Xu and J. Kawabata, Food Chem., 2007, 105, 628 CrossRef CAS.
- T. Fujisawa, H. Ikegami, K. Inoue, Y. Kawabata and T. Ogihara, Metab., Clin. Exp., 2005, 54, 387 CrossRef CAS PubMed.
- P. B. Fischer, G. B. Karlsson, T. D. Butters, R. A. Dwek and F. M. Platt, J. Virol., 1996, 70, 7143 CAS.
- P. B. Fischer, G. B. Karlsson, R. A. Dwek and F. M. Platt, J. Virol., 1996, 70, 7153 CAS.
- L. J. Scott and C. M. Spencer, Drugs, 2000, 59, 521 CrossRef CAS PubMed.
- H. Kings and M. Rewers, Diabetes Care, 1993, 16, 157 CrossRef.
- E. Rattanangkool, P. Kittikhunnatham, T. Damsud, S. Wacharasindhu and P. Phuwapraisirisan, Eur. J. Med. Chem., 2013, 66, 296 CrossRef CAS PubMed.
- A. Del Corso, M. Cappiello and U. Mura, Int. J. Biochem., 1994, 26, 745 CrossRef CAS PubMed.
- R. B. Freedman, FEBS Lett., 1979, 97, 201 CrossRef CAS PubMed.
- M. Gopalakrishnan, P. Sureshkumar, J. Thanusu and V. Kanagarajan, Pharm. Chem. J., 2008, 42, 271 CrossRef CAS.
- P. Yogeeswari, D. Banerjee, P. Bhat, A. Thomas, M. Srividya and D. Shriram, Eur. J. Med. Chem., 2011, 46, 106 CrossRef PubMed.
- N. Siddiqui and O. Singh, Indian J. Pharm. Sci., 2003, 65, 423 CAS.
- A. Chipeleme and J. Gut, Bioorg. Med. Chem., 2007, 17, 6434 CrossRef PubMed.
- C. Shipman, S. H. Smith, J. C. Drach and D. L. Klayman, Antimicrob. Agents Chemother., 1981, 19, 682 CrossRef CAS PubMed.
- (a) A. M. Thomas, A. D. Naik, M. Nethaji and A. R. Chakravarty, Inorg. Chim. Acta, 2004, 357, 2315 CrossRef CAS; (b) H. Beraldo and D. Gambino, Mini-Rev. Med. Chem., 2004, 4, 31 CrossRef CAS PubMed; (c) M. B. Ferrari, F. Bisceglie, G. G. Fava, G. Pelosi, P. Tarasconi and R. Albertini, et al., J. Inorg. Biochem., 2002, 89, 36 CrossRef; (d) H. S. Seleem, B. A. El-Shetary, S. M. E. Khalil, M. Mostafa and M. Shebl, J. Coord. Chem., 2005, 58, 479 CrossRef CAS.
- K. Zamani, K. Faghihi, S. Bagheri and M. Kalhor, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2004, 43, 2716 Search PubMed.
- C. M. Reis, D. S. Pereira, R. Oliveira, L. F. Kneipp and A. Echevarria, Molecules, 2011, 16, 10668 CrossRef PubMed.
- (a) S. Z. S. Farghaly, Master thesis, Assiut Univ., Faculty of Pharmacy, Assiut, Egypt, 1985; (b) H. Y. Hassan, Bull. Pharm. Sci., 1999, 22, 97 CAS.
- M. Rashid, A. Husain and R. Mishra, Eur. J. Med. Chem., 2012, 54, 855 CrossRef CAS PubMed.
- A. Husain, M. Rashid, R. Mishra, S. Parveen, D.-S. Shin and D. Kumar, Bioorg. Med. Chem. Lett., 2012, 22, 5438 CrossRef CAS PubMed.
- P. Puthiyapurayil, B. Poojary, C. Chikkanna and S. K. Buridipad, Eur. J. Med. Chem., 2012, 53, 203 CrossRef CAS PubMed.
- (a) N. K. N. Z. Abdullah, M. Taha, N. Ahmat, A. Wadood, N. H. Ismail, F. Rahim, M. Ali, N. Abdullah and K. M. Khan, Bioorg. Med. Chem., 2015, 23, 3119 CrossRef PubMed; (b) M. Taha, N. H. Ismail, S. Imran, M. Q. B. Rokei, S. M. Saad and K. M. Khan, Bioorg. Med. Chem., 2015, 23, 4155 CrossRef CAS PubMed.
- M. Taha, N. H. Ismail, S. Imran and K. M. Khan, Molbank, 2014, M826 CrossRef.
- (a) Z. Hadady, M. Toth and L. Somsak, ARKIVOC, 2004, 7, 140 Search PubMed; (b) M. Taha, N. H. Ismail, S. Imran, M. Selvaraj, A. Rahim, M. Ali, S. Siddiqui, F. Rahim and K. M. Khan, Bioorg. Med. Chem., 2015, 23, 7394 CrossRef CAS PubMed.
- M. J. Ahsan, J. G. Samy, H. Khalilullah, M. S. Nomani, P. Saraswat, R. Gaur and A. Singh, Bioorg. Med. Chem. Lett., 2011, 21, 7246 CrossRef CAS PubMed.
- F. Liu, X. Q. Luo, B. A. Song, P. S. Bhadury, S. Yang, L. H. Jin, W. Xue and D. Y. Hu, Bioorg. Med. Chem., 2008, 16, 3632 CrossRef CAS PubMed.
- V. Jakubkiene, M. M. Burbuliene, E. Udrenaite, V. Garaliene and P. Vainilavicius, Pharmazie, 2002, 57, 610 CAS.
- F. Rahim, K. Ullah, H. Ullah, A. Wadood, M. Taha, A. U. Rehman, I. Uddin, M. Ashraf, A. Shaukat, W. Rehman, S. Hussain and K. M. Khan, Bioorg. Chem., 2015, 58, 81 CrossRef CAS PubMed.
- M. Lödige and L. Hiersch, Int. J. Med. Chem., 2015, 458319 Search PubMed.
- (a) S. Imran, M. Taha, N. H. Ismail, S. M. Kashif, F. Rahim, W. Jamil and K. M. Khan, Chem. Biol. Drug Des., 2016, 87, 361–373 CrossRef CAS PubMed; (b) S. Imran, M. Taha, N. H. Ismail, S. M. Kashif, F. Rahim, W. Jamil, M. Hariono, M. Yusuf and H. Wahab, Eur. J. Med. Chem., 2015, 105, 156 CrossRef CAS PubMed.
- M. Naïm, S. Bhat, K. N. Rankin, S. Dennis, S. F. Chowdhury, I. Siddiqi, P. Drabik, T. Sulea, C. I. Bayly, A. Jakalian, E. O. Purisima and J. Chem, J. Chem. Inf. Model., 2007, 47, 122 CrossRef PubMed.
- M. Taha, N. H. Ismail, S. Imran, A. Wadood, F. Rahim, M. Ali and A. U. Rehman, MedChemComm, 2015, 6, 1826 RSC.
- S. B. Ferreira, A. C. R. Sodero, M. F. C. Cardoso, E. S. Lima, C. R. Kaiser, F. P. Silva and V. F. Ferreira, J. Med. Chem., 2010, 53, 2364 CrossRef CAS PubMed.
- J.-H. Park, S. Ko and H. Park, Bull. Korean Chem. Soc., 2008, 29, 921 CrossRef CAS.
- A. Roujeinikova, C. Raasch, S. Sedelnikova, W. Liebl and W. Rice, J. Mol. Biol., 2002, 321, 149 CrossRef CAS PubMed.
- L. R. Guerreiro, E. P. Carriero, L. Fernandes, T. A. Cardote, R. Moreira, A. T. Caldeira, R. C. Guedes and A. J. Burke, Bioorg. Med. Chem., 2013, 21, 1911 CrossRef CAS PubMed.
- M. Taha, N. H. Ismail, S. Lalani, M. Q. Fatmi, A.-T. Wahab, S. Siddiqui, K. M. Khan, S. Imran and M. I. Choudhary, Eur. J. Med. Chem., 2015, 92, 387 CrossRef CAS PubMed.
- T. A. Halgren, J. Comput. Chem., 1999, 20, 720 CrossRef CAS.
- J. Wang, P. Cieplak and P. A. Kollman, J. Comput. Chem., 2001, 21, 1049 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra28012e |
| This journal is © The Royal Society of Chemistry 2016 |