
Heteroarchitectured Ag–Bi2O3–ZnO as a bifunctional nanomaterial†
Subramanian Balachandranad,
Natarajan Prakashb and
Meenakshisundaram Swaminathan*c
aDepartment of Chemistry, Annamalai University, Annamalainagar – 608 002, India
bGraduate School of Science and Technology, Shizuoka University, 3-5-1 Johoku, Naka-ku, Hamamatsu 432-8011, Japan
cNanomaterials Laboratory, International Research Centre, Kalasalingam University, Krishnankoil-626126, Tamil Nadu, India. E-mail: chemres50@gmail.com; m.swaminathan@klu.ac.in; Fax: +91-4144-220572; Tel: +91-4144-220572
dBeijing National Laboratory for Molecular Sciences, Key Laboratory of Engineering Plastics, Institute of Chemistry, Chinese Academy of Sciences, Zhongguancun North First Street 2, Beijing 100190, P. R. China
First published on 9th February 2016
Abstract
Heteroarchitectured Ag–Bi2O3–ZnO was synthesized through a simple, cost effective, template free photodeposition-hydrothermal method. This composite material represents a prospective new class of novel catalysts wherein the improved light absorption behavior of a metal doped coupled oxide, Ag–Bi2O3–ZnO is anticipated to enhance with the observed benefits of a coupled system of Bi2O3–ZnO. Characterization of Ag–Bi2O3–ZnO revealed interesting optical and morphological characteristics. XRD patterns indicated that well-crystallized α-Bi2O3 was formed on the hexagonal wurtzite phase of ZnO along with Ag in the process. The HR-SEM images revealed the formation of organized intercrossed sheets and peanut shell like structure, which were composed of nanosheets and nanoparticles respectively. Elemental color mapping confirms the homogeneity of Zn, O, Ag and Bi in the catalyst. Ag–Bi2O3–ZnO has enhanced absorption in the UV and visible region when compared to Bi2O3 and ZnO. This catalyst exhibited enhanced photocatalytic activity for the degradation of Acid Red 1 (AR 1), Evens Blue (EB) and Acid Violet 7 (AV 7) under natural sunlight far exceeding those of the Bi2O3, Ag–ZnO, Ag–Bi2O3 and ZnO systems at neutral pH. Ag–Bi2O3–ZnO was found to be stable and reusable without appreciable loss of photocatalytic activity for up to five runs. Ag–Bi2O3–ZnO shows enhanced electrochemical methanol oxidation when compared to Ag–Bi2O3, Bi2O3–ZnO, Ag–ZnO and ZnO. Enhancement in methanol oxidation current demonstrates its high potential as an anode catalyst in direct methanol fuel cells.
Introduction
The design of efficient multifunctional catalysts will be a key to the present problems of environmental pollution and energy crisis. Semiconductor oxides afford a solution to these problems. ZnO, a semiconductor photocatalyst, has attracted intense research awareness in a broad range of applications, such as organic pollutant degradation, water splitting, and photovoltaic devices. ZnO was reported to be the most active semiconductor for the solar degradation of acid brown 14 among TiO2, ZnO, SnO2, ZrO2, α-Fe2O3, WO3 and CdS.1 The optical properties of ZnO can be tuned by controlling not only the size and shape of the nanocrystals but also the type and concentration of appropriate doping atoms.2–5Bismuth oxide (Bi2O3) is an important p-type semiconductor with four main crystallographic polymorphs denoted by α-, β-, γ-, and δ-Bi2O3. Bi2O3 with a band gap of 2.8 eV in the visible range has been widely used for various applications,6–10 and accounts for its ability to generate highly reactive species, such as O2−˙ and OH˙ radicals from water for mineralization of pollutants.11,12
But, the single-component UV light sensitive photocatalysts for environmental purification applications can only utilize photons below a wavelength of 450 nm, which represents only part of the solar light spectrum. One effective way to induce solar light absorption of a semiconductor is to couple it with another semiconductor having appropriate band structure. A large number of binary coupled semiconductor materials such as TiO2–SiO2,13 Bi2O3–ZnO,11 BiVO4–ZnO,14 Pr6O11–ZnO,15 Bi2S3–ZnO16 had been reported with enhanced photocatalytic activity. The surface adaption of semiconductors with noble metals (Ag, Pt, Au) has fascinated considerable attention especially in heterogeneous photocatalysis. This can reduce the recombination of the photogenerated electron/hole pairs and lengthen their lifetime through the conduction band electron trapping. Ag, as electron sinks, is well known due to the Schottky barrier at the metal–semiconductor interface.17 Nanocomposites such as TiO2–SiO2–Ag,18 TiO2–carbon–Ag,19 TiO2–graphene–Ag,20 TiO2–AgBr–Ag,21 and TiO2–graphene–Fe3O4 (ref. 22) have been synthesized. The development of new visible light-active photocatalysts is one of the most important topics in photocatalysis research.23
Methanol fuel cells (MFCs) are considered as significant power resource due to their low emissions and high energy efficiencies.24–28 However, the catalytic activity of high expensive of Pt and Pt-based electrocatalysts in MFCs diminish gradually due to chemisorption of carbonaceous intermediate products on these catalysts and this is the main barrier to the commercialization of direct methanol fuel cell (DMFC) technology.29–31 Thus, it is necessary to explore non-Pt catalysts to reduce costs and to improve the efficiency of the fuel cells. A significant challenge in the development of DMFC technology is the need for highly active anode catalysts for the methanol oxidation reaction.
Fabrication of heterostructured materials for multiple applications without using template or catalyst in a cost effective, simple methodology is a highly challenging job for chemists and material scientists. Herein, we report the preparation of Ag–Bi2O3–ZnO photocatalyst using simple photodeposition-hydrothermal method. The electrocatalytic activity of the catalyst for the electro-oxidation of methanol in alkaline media has been investigated.
Experiments
Reagents
Analar grade zinc nitrate hexahydrate, oxalic acid dihydrate, bismuth nitrate pentahydrate, silver nitrate, nitric acid, sodium hydroxide and absolute methanol were obtained from Himedia chemicals and used as such. Acid red 1 (molecular formula C18H13N3Na2O8S2 and molecular weight: 509.42), Evens blue (molecular formula C34H24N6Na4O14S4 and molecular weight: 960.8) and acid violet 7 (molecular formula C20H16N4Na2O9S2 and molecular weight: 566.47) from Colour Chem, Pondicherry and ZnO (surface area 5 m2 g−1, particle size 4.80 μm) from Merck chemicals were used as received. Deionized distilled water was employed throughout experiments.Preparation of Ag–Bi2O3–ZnO
0.97 g Bi(NO3)3·5H2O was dissolved in 10 mL 1.12 M nitric acid to avoid hydrolyzation of Bi3+ ions and before making up the solution to 100 mL, its pH was adjusted to 11 by the addition of small amount of 0.2 M NaOH. Then 2.0 mL of 0.1 M AgNO3 was quickly added to the above solution. This solution was mixed with 100 mL solution of 11.90 g of Zn (NO3)2·6H2O (0.4 M) in deionized water. Finally, 100 mL solution of oxalic acid dihydrate in deionized water (0.6 M) was introduced into the above solution drop wise with stirring and stirring was continued for 2 h to ensure complete precipitation of zinc oxalate and bismuth oxalate. The resulting suspension was stirred for 1.0 h and then UV irradiation was carried out using four parallel medium pressure mercury lamps emitting 365 nm wavelength for 3 h. The reaction mixture was transferred to the Teflon lined stainless steel autoclave for the hydrothermal treatment at 120 °C for 12 h. During the process the pressure was maintained at 18 psi. The final products were filtered, washed with deionized water and ethanol several times, and dried at 80 °C for 12 h in an oven. The mixed precipitate was calcined in air at 450 °C for 12 h, to get Ag–Bi2O3–ZnO. The preparation process is given in Scheme 1. This catalyst contains 9% Ag, 11.8% Bi2O3 and 79.2% ZnO. Catalysts with different percentages of Ag were prepared by a similar procedure with appropriate amount of AgNO3 solution. | ||
| Scheme 1 Schematic representation of preparation of Ag–Bi2O3–ZnO. | ||
Characterization
X-ray diffraction (XRD) patterns were recorded with a Siemens D5005 diffractometer using Cu Kα (k = 0.151418 nm) radiation. Maximum peak positions were compared with the standard files to identity the crystalline phase. The surface morphology of the Ag–Bi2O3–ZnO was studied using a field emission scanning electron microscope (FE-SEM) (Model ULTRA-55). EDS analysis was performed on gold coated samples with FE-SEM (Model ULTRA-55). HR-TEM images were taken from 200 kV ultra high resolution transmission electron microscope (JEOL-2010) having high resolution optical microscope (Leica microscope). Diffuse reflectance spectra were recorded with Shimadzu UV-2450. Photoluminescence spectra of the samples were recorded on a Perkin Elmer LS 55 fluorescence spectrometer. The excitation wavelength used to record PL was 320 nm. UV absorbance measurements were taken using Hitachi-U-2001 spectrometer.Photocatalytic experiments
All photocatalytic experiments were carried out under similar conditions on sunny days of April–May 2013 between 11 am and 2 pm. An open borosilicate glass tube of 50 mL capacity, 40 cm height and 20 mm diameter was used as the reaction vessel. 50 mL of dye solution (5 × 10−4 M) with 200 mg of Ag–Bi2O3–ZnO was magnetically stirred in the dark for 30 min to attain adsorption–desorption equilibrium between the dye and Ag–Bi2O3–ZnO. After dark adsorption the first sample was taken. Solar irradiation was carried out in the open-air condition. The suspension was continuously aerated (airflow rate = 8.1 mL s−1) by a pump to provide oxygen and for the complete mixing of reaction solution. During the illumination time no volatility of the solvent was observed. At specific time intervals, 2 mL of the sample was withdrawn and centrifuged to separate the catalyst. 1 mL of the centrifugate was diluted to 10 mL and its absorbance was measured at 306, 314 and 285 nm for AR 1, EB and AV 7 dyes respectively. The absorbance at 306, 314 and 285 nm represent the aromatic content of AR 1, EB and AV 7 respectively and its decrease indicates extent of dye degradation. Solar light intensity was measured for every 30 min and the average light intensity over the duration of each experiment was calculated. The sensor was always set in the position of maximum intensity. The intensity of solar light was measured using LT Lutron LX-10/A Digital Lux meter and the intensity was 1250 × 100 ± 100Lux. The intensity was nearly constant during the experiments.Electrochemical studies
Cyclic voltammetry (CV) was performed with a CHI660 electrochemical workstation (CH Instruments, USA). Cyclic voltammetry was carried out with three electrodes, Ag/AgCl electrode as the reference electrode, a counter electrode (platinum wire) and unmodified or modified glassy carbon electrode (GCE) as the working electrode. GCEs were polished before the experiments in sequence with 1, 0.30 and 0.05 micron aluminium/water slurry on micro cloth pads, followed by careful cleaning in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 HNO3–H2O (v/v), ethanol, and water via ultra-sonication. The working electrode was prepared by spraying the catalysts dispersed in isopropyl alcohol. Cyclic voltammograms were obtained between +1.0 and −0.1 V at a scan rate of 50 mV s−1. All potentials are reported with respect to the reversible hydrogen electrode. The CO stripping voltammetry curves were obtained after adsorption of CO on catalysts at −0.2 V. To observe methanol electro-oxidation, 0.5 M methanol solution in 0.5 M NaOH was used.
:1 HNO3–H2O (v/v), ethanol, and water via ultra-sonication. The working electrode was prepared by spraying the catalysts dispersed in isopropyl alcohol. Cyclic voltammograms were obtained between +1.0 and −0.1 V at a scan rate of 50 mV s−1. All potentials are reported with respect to the reversible hydrogen electrode. The CO stripping voltammetry curves were obtained after adsorption of CO on catalysts at −0.2 V. To observe methanol electro-oxidation, 0.5 M methanol solution in 0.5 M NaOH was used.
Results and discussion
Preliminary study on the degradation of three azo dyes AR 1, EB and AV 7 with different wt% of Ag in Ag–Bi2O3–ZnO catalysts was carried out. The percentages of AR1 degradation with 3, 6, 9, 12 and 15% of Ag were found to be 67, 70, 75, 72 and 69% respectively for 60 minutes irradiation and the percentage of error is +1, or −1 in the consecutive measurements. The maximum efficiency was observed with 9 wt% Ag loaded catalyst. Similar results were obtained with EB and AV 7 degradation. Hence this 9 wt% Ag loaded Bi2O3–ZnO was characterized and used for further experiments. Fig. 1a–d, show the typical X-ray diffraction pattern of the prepared ZnO, 3 wt%, 6 wt%, 9 wt% of Ag in Bi2O3–ZnO nanocatalysts respectively. The 2θ values of prepared ZnO at 31.77, 34.49, 36.24, 56.60, 62.85, 66.38, 67.94, 69.08, 72.50 and 76.93 correspond to (1 0 0), (0 0 2), (1 0 1), (1 1 0), (1 0 3), (2 2 0), (1 1 2), (2 0 1), (0 0 4) and (2 0 2) planes of wurtzite ZnO (Fig. 1a). The hexagonal wurtzite structure of ZnO consists of alternating planes of tetrahedrally coordinated oxygen and zinc atoms arranged along the c-axis. This anisotropy results in the spontaneous polarization of certain planes, e.g., {001} or {101}, whereas other planes are nonpolar, e.g., {100}.31 The new peaks observed at 27.9, 30.11, 33.7, 46.6, 47.1 correspond to (121), (012), (−122), (041), (−104) planes of α-Bi2O3 (JCPDS # 71-2274)32 in Bi2O3–ZnO nanocatalysts (Fig. 1b–d). The crystallographic phase of the prepared Ag–Bi2O3–ZnO is confirmed as monoclinic α-Bi2O3 form present in hexagonal wurtzite ZnO base material. The relatively high intensity of the (101) peak is indicative of anisotropic growth and implies a preferred orientation of the crystallites. The peaks at 2θ = 38.2 and 46.23° correspond to (111) and (200) planes of face-centered-cubic (fcc) lattice of metallic Ag (JCPDS file no. 04-0783).33 The increase in the concentration of Ag from 3 wt% to 9 wt% in Ag–Bi2O3–ZnO increases the intensity of Ag peaks at 2θ = 38.2 and 46.23°. | ||
| Fig. 1 XRD patterns of (a) prepared ZnO and (b) 3 wt% Ag–Bi2O3–ZnO, (c) 6 wt% Ag–Bi2O3–ZnO and (d) 9 wt% Ag–Bi2O3–ZnO. | ||
The Field emission scanning electron microscope (FE-SEM) images of 9 wt% Ag–Bi2O3–ZnO at three different magnifications are shown in Fig. 2. FE-SEM images show that Ag–Bi2O3–ZnO has an ordered mixture of heterostructured intercrossed micro sheets and nano particles (Fig. 2a–c) and the size of the particles are in the range of 20–35 nm (Fig. 2c). The nanoparticles are highlighted in Fig. 2a by green circle. The hydrothermal method reported here is a simple process without rigorous conditions and hence it is a low-cost and convenient method to prepare nano sheet shaped Ag–Bi2O3–ZnO.
 | ||
| Fig. 2 FE-SEM images of 9 wt% Ag–Bi2O3–ZnO at different magnification (a) 2 μm (nano particles are highlighted by green circle), (b) 1 μm and (c) 50 nm. | ||
The surface morphology was further investigated by HR-SEM images (Fig. 3a–c). These images confirm that Ag–Bi2O3–ZnO is composed of nanoparticles and nanosheets. During the hydrothermal process nanoparticles are self-assembled to form a sheet like structures (Fig. 3). Nanosheets are attached with other nanosheets and give an intercrossed sheet like structure (Fig. 3c). This heterostructure having a large number of cavities on surfaces may enhance the dye adsorption on Ag–Bi2O3–ZnO and favor for active dye degradation.
 | ||
| Fig. 3 HR-SEM images of 9 wt% Ag–Bi2O3–ZnO at different magnification (a) 300 nm, (b) 1 μm and (c) 1 μm. | ||
To confirm the distribution of Zn, O, Bi and Ag in the catalyst, elemental mapping of FE-SEM was carried out. Fig. 4a–d show the elemental mapping for zinc, oxygen, bismuth and silver respectively. It is evident from the Fig. 4a and b that Zn and O are higher in density. There is a homogenous distribution of Bi and Ag at lower concentration (Fig. 4c and d). Thus elemental mapping shows that the catalyst is composed of Zn, O, Bi and Ag. This also indicates the purity of the catalyst Ag–Bi2O3–ZnO.
 | ||
| Fig. 4 FESEM elemental colour mapping of 9 wt% Ag–Bi2O3–ZnO (a) Zn, (b) O, (c) Bi and (d) Ag. | ||
Typical TEM images of the Ag–Bi2O3–ZnO are shown in Fig. 5a–c. This exhibits assembled nanoparticles with a large number of sheets. TEM images show a sheet with sphere like structure of irregular morphology. Fig. 5d shows the high-resolution TEM image of the Ag–Bi2O3–ZnO. The lattice spacing of 0.328 nm corresponds to the interlayer spacing of the (012) plane of the α-Bi2O3 phase and lattice spacing of 0.256 nm is for (101) planes of wurtzite ZnO.33 The lattice fringes with 0.236 nm spacing were assigned to the Ag (111) plane. The results match exactly with XRD pattern discussed earlier.
 | ||
| Fig. 5 TEM images of 9 wt% Ag–Bi2O3–ZnO (a), (b), (c) particle distribution and (d) HR-TEM image (lattice fringes). | ||
In order to find out the presence of elements and to determine their valance states in Ag–Bi2O3–ZnO, XPS study was carried out. Survey spectrum in Fig. 6a shows the presence of Zn, O Bi and Ag. Carbon 1s peaks of the Ag–Bi2O3–ZnO are ascribed to adventitious hydrocarbon from XPS instrument itself. In Fig. 6b, the O 1s profile is asymmetric and can be fitted to two symmetrical peaks α and β locating at 530.2 and 532.2 eV, respectively, indicating two different kinds of O species in the sample. The peaks α and β should be associated with the lattice oxygen (OL) of ZnO and chemisorbed oxygen (OH) respectively.34 Fig. 6c presents the XPS spectra of Zn 2p, and the peak positions of Zn 2p1/2 and Zn 2p3/2 locate at 1045.1 eV and 1021.9 eV. Comparing the peak positions to those in the Handbook of X-Ray Photoelectron Spectroscopy, we can conclude that: (i) Zn is in the state of Zn2+ and (ii) the whole XPS spectra have a downward shift of 0.5 eV since the standard peak position of Zn 2p3/2 is at 1022.4 eV.35 Two signals ascribed to Bi 4f7/2 and Bi 4f5/2 at binding energies of 158.6 and 164.1 eV are observed in Fig. 6d. There are two peaks locating at 368.4 and 374.5 eV, which are attributed to Ag 3d5/2 and Ag 3d3/2, respectively (Fig. 6e). According to Zhang et al., these two peaks can be attributed to metallic Ag (Ag0).36 The binding energy peaks of Ag 3d5/2 for the Ag/ZnO sample shifts upward when compared with the corresponding value of the synthesized pure metallic Ag (binding energy value of Ag0 is 368.2). The shift must be due to the strong interaction between Ag and ZnO.
 | ||
| Fig. 6 XPS analysis of 9 wt% Ag–Bi2O3–ZnO (a) survey spectrum, (b) O 1s, (c) Zn 2p, (d) Bi 4f and (e) Ag 3d. | ||
Optical studies
The UV-visible diffuse reflectance spectra of the Ag–Bi2O3–ZnO nanocomposite and ZnO were measured to determine their light absorption characteristics (Fig. 7). The wavelength distribution of the absorbed light is a significant property of photocatalysts. Ag–Bi2O3–ZnO nanocomposite has higher UV and visible light absorbance, as indicated by the diffuse reflectance spectrum in absorption mode (Fig. 7b). The Ag–Bi2O3–ZnO catalyst shows the enhanced absorption in the visible region. The extended absorption of the Ag–Bi2O3–ZnO nanocomposite in the visible region, is due to the typical surface plasmon band exhibited by the AgNPs.37 | ||
| Fig. 7 Diffuse reflectance spectra (a) prepared ZnO and (b) 9 wt% Ag–Bi2O3–ZnO. | ||
Photoluminescence (PL) measurements were performed to determine the charge recombination efficiency of the Ag–Bi2O3–ZnO nanocomposite and ZnO because the photocatalytic activity is related closely to the recombination rate of photoexcited electrons and holes. Fig. 8 shows the PL spectra of the Ag–Bi2O3–ZnO (Fig. 8b) nanocomposite and ZnO (Fig. 8a). Both spectra mainly consist of two emission bands: a band at 416 nm (2.98 eV) and a blue-green band at 485 nm (2.56 eV). The strong UV emission at 416 nm corresponds to the electron–hole recombination.38–41 Reduction of PL intensity at 416 nm of Ag–Bi2O3–ZnO when compared to prepared ZnO indicates the suppression of recombination of the photogenerated electron–hole pair. The emission intensity of the Ag–Bi2O3–ZnO nanocomposite is lower than that of ZnO, suggesting that the anchoring of Ag, and Bi2O3 quench the fluorescence from the ZnO nanoparticles and prolong the electron–hole pair lifetime enhancing the photocatalytic activity of Ag–Bi2O3–ZnO.
 | ||
| Fig. 8 Photoluminescence spectra of (a) prepared ZnO and (b) 9 wt% Ag–Bi2O3–ZnO. | ||
Photocatalytic activity of the heterostructured Ag–Bi2O3–ZnO
Photocatalytic degradation of Acid Red 1 (AR 1) under different conditions is shown in Fig. 9. Dye is resistant to self-photolysis and for the same experiment with Ag–Bi2O3–ZnO in dark, a small decrease (8%) in dye concentration was observed due to the adsorption of dye on the catalyst. AR 1 undergoes 98.6% degradation in the presence of Ag–Bi2O3–ZnO under natural sunlight in 90 min. But, prepared Ag–Bi2O3, Ag–ZnO, Bi2O3 and ZnO produced 74, 85, 62 and 81% degradations, respectively in 90 min. This shows that Ag–Bi2O3–ZnO is most efficient in AR 1 degradation than other photocatalysts (Fig. 9). Based on the results, it is obvious that the higher photocatalytic activity of Ag–Bi2O3–ZnO is due to the Ag and Bi2O3 dopants. To test the efficiency of the catalyst on the degradations of other dyes, we had carried out the experiments on the degradation of EB and AV 7 under the same conditions. The degradation of three azo dyes AR1, EB and AV 7 at different irradiation times are shown in Fig. S2, S3 and S4† respectively. There is no significant change in UV-vis spectra of dyes during irradiation, but the intensity decrease gradually during degradation. This reveals that the intermediates do not absorb at analytical wavelengths. Similar results were observed in the photocatalytic degradation of azo dyes.14,15 Fig. S5† shows the percentages of degradation of EB and AV 7 along with AR1 using Ag–Bi2O3–ZnO at different irradiation times. All the three dyes undergo almost complete degradation in 90 min. The results reveal that this catalyst is efficient in the degradation of azo dyes. In the case of EB dye, 44% of dye was absorbed by Ag–Bi2O3–ZnO. Experiments were conducted to find out whether the adsorbed dye molecules had been completely degraded by the reported procedure.42 The results revealed that the adsorbed dye underwent complete degradation in 90 minutes.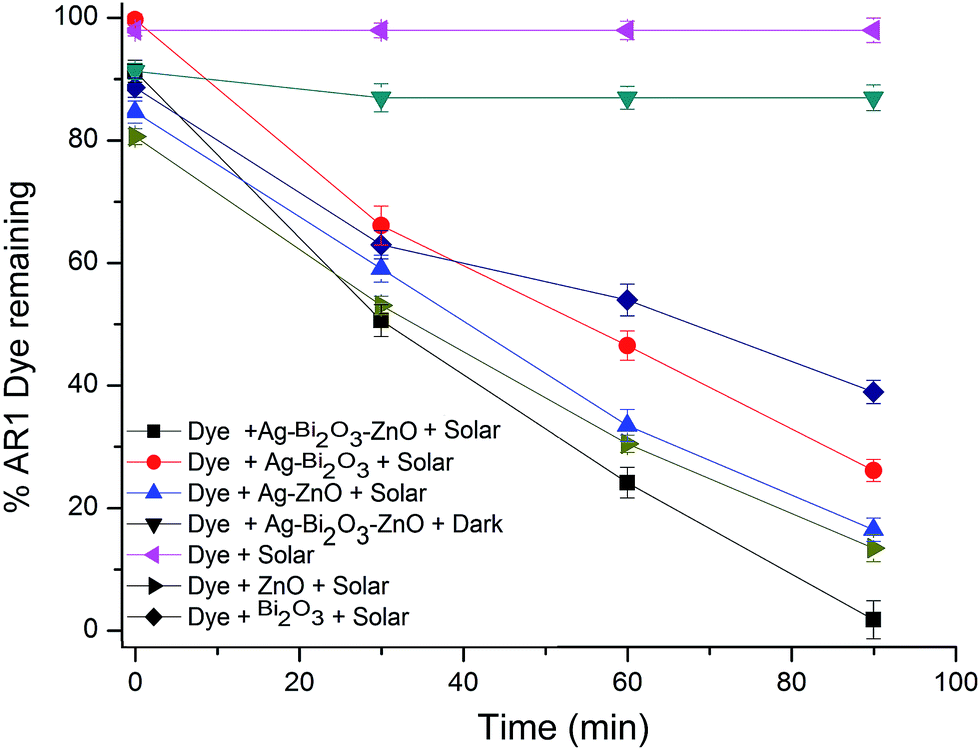 | ||
| Fig. 9 Primary analysis of Ag–Bi2O3–ZnO: AR 1 dye concentration = 5 × 10−4 M, catalyst suspended = 4 g L−1, pH = 7, airflow rate = 8.1 mL s−1, Isolar = 1250 × 100Lux ± 100. | ||
The percentage of degradation in the solar process is affected by variables such as the pH and catalyst loading. The effects of these variables were studied using the azo dyes (AR 1, EB and AV 7) with Ag–Bi2O3–ZnO. The acid–base property of the metal oxide surfaces can have considerable implications on their photocatalytic activity. Percentages of degradation at different pH from 3–11 for three azo dyes are shown in Fig. 10. It is observed that the degradation rate increases with an increase in pH up to 7 and then decreases. After 90 min of irradiation, the percentages of AR 1 degradation are 53, 71, 96, 78 and 62 at pH 3, 5, 7, 9 and 11, respectively. The optimum pH is found to be 7 for AR 1 degradation and a similar trend is observed for EB and AV 7 dyes. Low removal efficiency at the acidic pH range may be due to the dissolution of ZnO in Ag–Bi2O3–ZnO. To find out the reason for the effect of pH on degradation efficiency, zero point charge (ZPC) of the catalyst was determined by the potentiometric titration method.43 Zero point charge of Ag–Bi2O3–ZnO is found to be 7.1, which is less than ZPC of ZnO (9). When the pH is above ZPC, the surface charge density of the catalyst becomes negative. This affects the adsorption of dye molecules, which exist anions in the whole range of pH studied. Hence the degradation efficiency is low at pH 9 and 11.
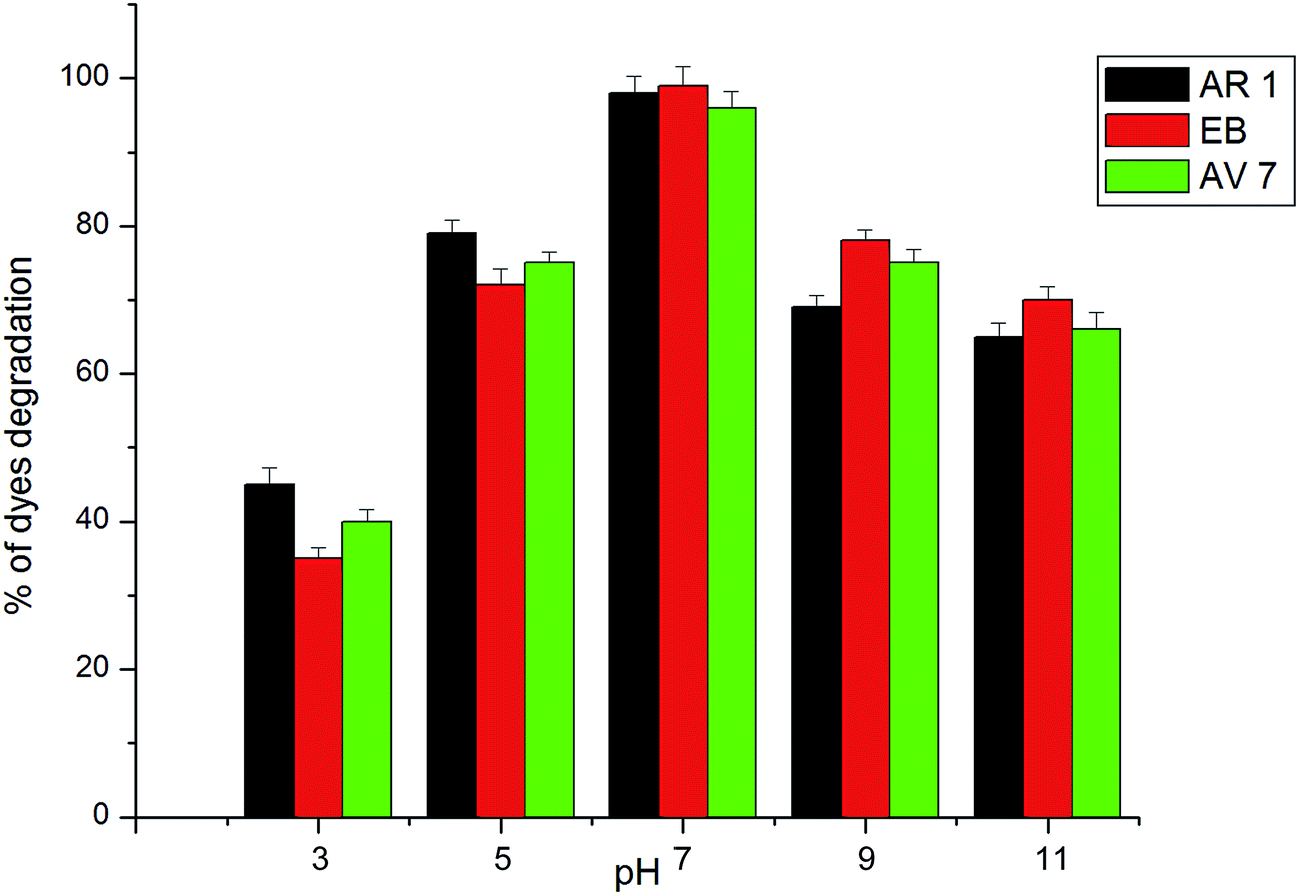 | ||
| Fig. 10 Effect of solution pH; AR 1 dye concentration = 5 × 10−4 M, catalyst suspended = 4 g L−1, airflow rate = 8.1 mL s−1, Isolar = 1250 × 100Lux ± 100, (b) EB dye concentration = 5 × 10−4 M, catalyst suspended = 4 g L−1, airflow rate = 8.1 mL s−1, Isolar = 1250 × 100Lux ± 100 lx, (c) primary analysis: AV 7 dye concentration = 5 × 10−4 M, catalyst suspended = 4 g L−1, airflow rate = 8.1 mL s−1, Isolar = 1250 × 100Lux ± 100. | ||
The reusability of Ag–Bi2O3–ZnO was tested for the degradation of azo dyes under identical reaction conditions. After complete degradation, the catalyst was separated and washed with large amount of deionized water. The recovered catalyst was dried in hot air oven at 100 °C for 90 min and used for a second run. Fig. S6† shows the results of dyes degradation for five runs. Ag–Bi2O3–ZnO exhibits remarkable photostability as the AR 1 degradation percentages are 98, 96, 94.0, 94 and 94 for 90 min in the first, second, third, fourth and fifth run respectively. There is no significant change in the degradation efficiency of Ag–Bi2O3–ZnO after third run. After completion of degradation reaction, the solution was tested for Bi3+ leaching with sodium sulfide. There is no precipitation of bismuth sulfide (black color). As there is no leaching of Bi3+ this catalyst is non-toxic for the wastewater treatment.
Mechanism of degradation
Mechanism of the photocatalytic activity of the Ag–Bi2O3–ZnO composite is proposed in Scheme 2. Three main reasons for the increase in the photocatalytic efficiency of metal doped coupled oxide: (i) visible absorbance of Ag–Bi2O3–ZnO increased when compared to prepared ZnO, (ii) band energy levels of Bi2O3 and ZnO are suitable for charge separation and (iii) Ag metal can act as electron trap. The conduction band edges of ZnO and Bi2O3 are situated at −0.38 and +0.32 eV, while the valence band edges of Bi2O3 and ZnO are at +2.78 eV and 2.84 eV respectively as shown in Scheme 2. The CB of Bi2O3 is much more positive (+0.7 eV) than ZnO whereas VB of Bi2O3 is slightly more negative (−0.06 eV) than ZnO. Hence there is a greater tendency for the flow of electrons from ZnO to Bi2O3. As there is no significant difference in VB levels, the tendency for the holes to flow in to Bi2O3 will be less. This makes the charge separation effective. The enhanced charge separation is also revealed by the reduction of PL intensity of the catalyst when compared to ZnO (Fig. 8). Further the separation of photogenerated electron–hole may be due to the formation of heterojunction between ZnO and Bi2O3, leading to enhancement of photocatalytic activity. In addition to this ‘Ag’ metal can trap the electron from ZnO and Bi2O3, inhibiting the recombination of electrons and holes in Ag–Bi2O3–ZnO. Holes remained on ZnO valence band and electrons concentrated on Bi2O3 conduction band, enabled this type of three component system with strong oxidation and reduction capabilities to generate superoxide (O2˙−), and hydroxyl radicals (OH˙) for the dye degradation.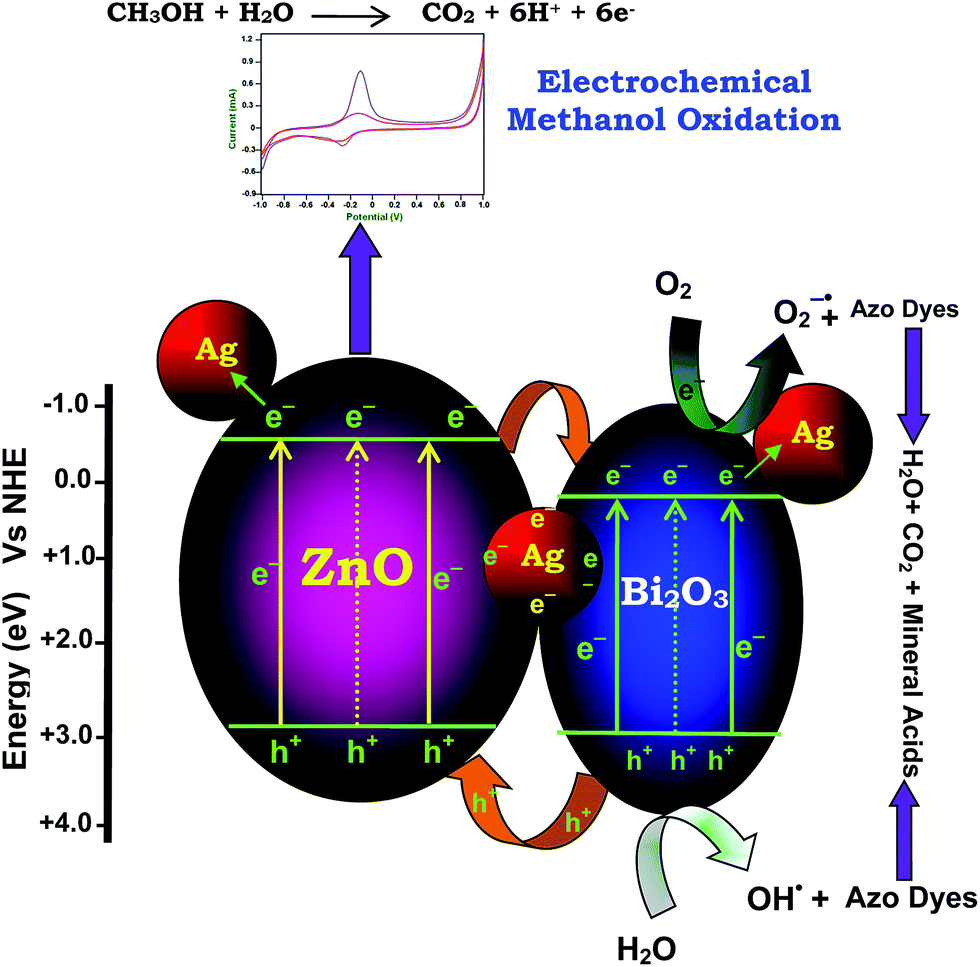 | ||
| Scheme 2 Dye degradation mechanism. | ||
In the case of EB dye, a dye-sensitized mechanism is also possible for the degradation. We carried out the degradation of EB with 365 nm UV light (IUV = 1.381 × 10−6 Einstein L−1 s−1) under the same conditions used for natural sun light. It was found that EB underwent 76.3% degradation with UV light, but under the same conditions 90% degradation occurred with solar light in 75 min. The higher efficiency in solar light indicates the presence of a dye-sensitized mechanism in addition to Ag–Bi2O3–ZnO sensitization. This occurs when more dye molecules are adsorbed on the semiconductor surface (42.6% EB adsorption in dark). Dye is sensitized by solar irradiation and loses an electron (eqn (1)) and this electron is transferred to the conduction band of ZnO. This subsequently increases electron transfer to the adsorbed oxygen producing superoxide radicals (eqn (2)). Dye molecules are degraded by the superoxide radicals produced by the dye sensitization mechanism (eqn (3)). Further, to prove the dye-sensitized mechanism, we had also carried out an experiment for degradation of colorless 4-nitrophenol by Ag–Bi2O3–ZnO with UV and solar light. We found that degradation of colorless 4-nitrophenol (absorbing only in the UV region) was more efficient in UV light (81.9%) than in solar light (54.2%) in 75 min under the same conditions, indicating the presence of only catalyst sensitized mechanism in the degradation of 4-nitrophenol. This confirms the presence of dye-sensitized mechanism for degradation of EB dye.
| EB dye + hν → EB dye+˙ + ecb− | (1) |
| ecb− + O2 → O2˙− | (2) |
| EB dye+˙ + O2/O2˙− → degradation products | (3) |
Electrocatalytic methanol oxidation
The structure of the electrode greatly influences its electrocatalytic activity. It has been proved that the electro-oxidation of methanol is a surface-sensitive reaction.29,30 Pt wire and Ag/AgCl electrodes were used as the counter and reference electrodes, respectively. Fig. 11 shows the cyclic voltammograms (CVs) of glass carbon electrode coated with prepared ZnO (a), Ag–Bi2O3–ZnO (b) taken in N2 saturated 0.5 M NaOH solution at a scan rate of 50 mV s−1. The CVs (a and b) show typical potential regions for hydrogen adsorption/desorption and the formation/reduction of surfaces of metal oxide or metal–OHads ZnO and Ag–Bi2O3–ZnO. | ||
| Fig. 11 Cyclic voltammogram of (a) prepared ZnO and (b) 9 wt% Ag–Bi2O3–ZnO in N2 and saturated 0.5 M NaOH + 0.5 M CH3OH solution at a scan rate of 50 mV s−1 at 25 °C. | ||
The smaller size and good dispersion of Ag–Bi2O3–ZnO modified GCE are favorable for the oxidation of methanol. Two peaks of methanol oxidation under anodic condition are clearly observed (Fig. 11a and b). The oxidation peak in the forward scan corresponds to the oxidation of freshly chemisorbed species coming from methanol adsorption and the reverse oxidation peak is primarily associated with removal of carbonaceous species not completely oxidized in the forward scan. The magnitude of the peak current on the forward scan indicated the electrocatalytic activity of the coated catalysts for methanol oxidation. It is clearly seen that Ag–Bi2O3–ZnO catalyst exhibits a current density of 3.13 mA, which is about 3 times higher than that of and ZnO (1.12 mA).
It is well recognized that the stability of catalysts is essential for the application of fuel cells. However, redox reactions on the surface of catalysts often cause inactivation of catalyst. To find out the electrocatalytic stability of the catalysts under continuous operating conditions, long-term chronoamperometric experiments were carried out in 0.5 M NaOH + 0.5 M methanol solution.44 Fig. S7† shows the curves of current densities versus time recorded at − 0.1 V for 1000 s. All these catalysts initially show a rapid decrease in current for the oxidation of methanol, and then a relatively steady current is achieved. The rapid current decay showed the poisoning of the electrocatalysts, likely due to the formation of intermediate and some poisoning species during the methanol oxidation reaction.45,46 It is found that the current decay for the reaction on the Ag–Bi2O3–ZnO (curve b) is significantly less than on the prepared ZnO (curve a). This suggests that the Ag–Bi2O3 loading plays a critical role in promoting the methanol oxidation, which could be attributed to the intrinsic electron transfer characteristic of Ag–Bi2O3–ZnO. This reveals the higher stability of the Ag–Bi2O3–ZnO catalyst for methanol oxidation than ZnO.
Conclusions
Heteroarchitectured Ag–Bi2O3–ZnO semiconductor photocatalyst was synthesized by the photodeposition-hydrothermal method and characterized. The HR-SEM images revealed the formation of well-organized intercrossed sheets composed of nanosheets and nanoparticles. Elemental color mapping confirms the homogeneity of Zn, O, Ag and Bi in the catalyst. Ag–Bi2O3–ZnO has a nanoparticles-intercrossed sheet like morphology and with high porosity. The Ag–Bi2O3–ZnO has increased absorption in the UV and visible region. Ag–Bi2O3–ZnO was more efficient in dye degradation than the prepared ZnO, Ag–Bi2O3, and TiO2–P25 under natural sunlight. The optimum pH for the efficient removal of dye was found to be 7. This hydrothermal route for the fabrication of Ag–Bi2O3–ZnO is effective, fast, convenient, and eco-friendly. A mechanism for high charge separation efficiency is proposed. This catalyst was found to stable and reusable. Electrocatalytic activity of Ag–Bi2O3–ZnO nanocomposite exhibited enhanced current production by methanol oxidation under room temperature.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
We are grateful to the Council of Scientific and Industrial Research, New Delhi, for the financial support through Research Grant no. 21 (0799)/10/EMR-II. One of the authors Dr S. Balachandran is thankful to the UGC Networking Resource Centre, University of Hyderabad for providing characterization facility and to Dr Tushar Jana, School of Chemistry, University of Hyderabad for the laboratory facility. Authors are grateful to Dr P. V. Satyam, Institute of Physics, Bhubaneswar for using HR-TEM facility.Notes and references
- S. Sakthivel, B. Neppolian, M. V. Shankar, B. Arabindoo, M. Palanichamy and V. Murugesan, Sol. Energy Mater. Sol. Cells, 2003, 77, 65–82 CrossRef CAS.
- C. G. van de Walle, Phys. Rev. Lett., 2000, 85, 1012–1015 CrossRef CAS PubMed.
- K. T. Ramakrishna, H. Gopalaswamy and P. J. Reddy, J. Cryst. Growth, 2000, 210, 516–520 CrossRef.
- P. Cao, D. X. Zhao, J. Y Zhang, D. Z. Shen, Y. M. Lu, B. Yao, B. H. Li, Y. Bai and X. W. Fan, Appl. Surf. Sci., 2008, 254, 2900–2904 CrossRef CAS.
- J. B. Yi, C. C. Lim, G. Z. Xing, H. M. Fan, L. H. Van, S. L. Huang, K. S. Yang, X. L. Huang, X. B. Qin, B. Y. Wang, T. Wu, L. Wang, H. T. Zhang, X. Y. Gao, T. Liu, A. T. S. Wee, Y. P. Feng and J. Ding, Phys. Rev. Lett., 2010, 104, 137201–137204 CrossRef CAS PubMed.
- A. Cabot, A. Marsal, J. Arbiol and J. R. Morante, Sens. Actuators, B, 2004, 99, 74–89 CrossRef CAS.
- L. Leontie, M. Caraman, M. Delibas and G. I. Rusu, Mater. Res. Bull., 2001, 36, 1629–1637 CrossRef CAS.
- D. Kulkarni and I. E. Wachs, Appl. Catal., A, 2002, 237, 121–137 CrossRef CAS.
- J. Switzer, M. Shumsky and E. Bohannan, Science, 1999, 284, 293–296 CrossRef CAS PubMed.
- C. E. Mohn, S. Stolen, S. T. Norberg and S. Hull, Phys. Rev. Lett., 2009, 102, 155502–155506 CrossRef PubMed.
- S. Balachandran and M. Swaminathan, J. Phys. Chem. C, 2012, 116, 26306–26312 CAS.
- M. Muruganandham, R. Amutha, G. Juan Lee, S. Han Hsieh, J. J. Wu and M. Sillanpaa, J. Phys. Chem. C, 2012, 116, 12906–12915 CAS.
- D. Yang, C. Chen, Z. Zheng, H. Liu, E. R. Waclawik, Z. Yan, Y. Huang, H. Zhang, J. Zhao and H. Zhu, Energy Environ. Sci., 2011, 4, 2279–2287 CAS.
- S. Balachandran, N. Prakash, K. Thirumalai, M. Muruganandham, M. Sillanpaa and M. Swaminathan, Ind. Eng. Chem. Res., 2014, 53, 8346–8356 CrossRef CAS.
- S. Balachandran, K. Thirumalai and M. Swaminathan, RSC Adv., 2014, 4, 27642–27653 RSC.
- S. Balachandran and M. Swaminathan, Dalton Trans., 2013, 42, 5338–5347 RSC.
- Y. Liang, H. Wang, S. H. Casalongue, Z. Chen and H. Dai, Nano Res., 2010, 3, 701–705 CrossRef CAS.
- C. Ren, B. Yang, M. Wu, J. Xu, Z. Fu, Y. lv, T. Guo, Y. Zhao and C. Zhu, J. Hazard. Mater., 2010, 182, 123–129 CrossRef CAS PubMed.
- (a) Q. Chen, H. Shi, W. Shi, Y. Xu and D. Wu, Catal. Sci. Technol., 2012, 2, 1213–1220 RSC; (b) C. Liu, D. Yang, Y. Jiao, Y. Tian, Y. Wang and Z. Jiang, ACS Appl. Mater. Interfaces, 2013, 5, 3824–3832 CrossRef CAS PubMed.
- P. Zhang, C. Shao, Z. Zhang, M. Zhang, J. Mu, Z. Guo, Y. Sun and Y. Liu, J. Mater. Chem., 2011, 21, 17746–17753 RSC.
- Y. Wen, H. Ding and Y. Shan, Nanoscale, 2011, 3, 4411–4417 RSC.
- G. Tian, Y. Chen, H. L. Bao, X. Meng, K. Pan, W. Zhou, C. Tian, J. Q. Wang and H. Fu, J. Mater. Chem., 2012, 22, 2081–2088 RSC.
- Y. Lin, Z. Geng, H. Cai, L. Ma, J. Chen, J. Zeng, N. Pan and X. Wang, Eur. J. Inorg. Chem., 2012, 2012, 4439–4444 CrossRef CAS.
- Z. L. Liu, B. Zhao, C. L. Guo, Y. J. Sun, Y. Shi, H. B. Yang and Z. Li, J. Colloid Interface Sci., 2010, 351, 233–238 CrossRef CAS PubMed.
- Y. M. Li, L. H. Tang and J. H. Li, Electrochem. Commun., 2009, 11, 846–849 CrossRef CAS.
- R. Ganesan and J. S. Lee, Angew. Chem., Int. Ed., 2005, 44, 6557–6560 CrossRef CAS PubMed.
- Q. F. Yi, F. J. Niu, L. H. Song, X. P. Liu and H. D. Nie, Electroanalysis, 2011, 23, 2232–2240 CrossRef CAS.
- Z. Liu, X. Zhang and L. Hong, Electrochem. Commun., 2009, 11, 925–928 CrossRef CAS.
- R. M. A. Hameed and K. M. E. Khatib, Int. J. Hydrogen Energy, 2010, 35, 2517–2529 CrossRef.
- Y. Zhang, G. Chang, S. Liu, J. Tian, L. Wang, W. Lu, X. Qin and X. Sun, Catal. Sci. Technol., 2011, 1, 1636–1640 CAS.
- Z. Y. Jiang, Q. Kuang, Z. X. Xie and L. S. Zheng, Adv. Funct. Mater., 2010, 20, 3634–3645 CrossRef CAS.
- N. Pugazhenthiran, P. Sathishkumar, S. Murugesan and S. Anandan, Chem. Eng. J., 2011, 168, 1227–1233 CrossRef CAS.
- D. Lin, H. Wu, R. Zhang and W. Pan, Chem. Mater., 2009, 21, 3479–3484 CrossRef CAS.
- J. J. F. Moudler, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, Perkin-Elmer Corp, Eden Prairie, MN, 1995, p. 127 Search PubMed.
- C. D. Wagner, W. M. Riggs, L. E. Davis and J. F. Moulder, Handbook of X-ray Photoelectron Spectroscopy, Perkin Elmer, Eden Prairie, 1979, p. 81 Search PubMed.
- L. Kuai, B. Geng, X. Chen, Y. Zhao and Y. Luo, Langmuir, 2010, 26, 18723–18727 CrossRef CAS PubMed.
- Z. Zhan, J. An, H. Zhang, R. V. Hansen and L. Zheng, ACS Appl. Mater. Interfaces, 2014, 6, 1139–1144 CAS.
- V. Stikant and D. R. Clarke, J. Appl. Phys., 1998, 83, 5447–5451 CrossRef.
- S. C. Lyu, Y. Zhang, H. Ruh, H. Lee, H. Shim, E. Suh and C. J. Lee, Chem. Phys. Lett., 2002, 363, 134–138 CrossRef CAS.
- L. Bergman, X. B. Chen, J. L. Morrison, J. Huso and A. P. Purdy, J. Appl. Phys., 2004, 96, 675–682 CrossRef CAS.
- J. Wang and L. Gao, Solid State Commun., 2004, 132, 269–271 CrossRef CAS.
- R. Velmurugan, B. Krishnakumar, B. Subash and M. Swaminathan, Sol. Energy Mater. Sol. Cells, 2013, 108, 205–212 CrossRef CAS.
- S. Subramanian, J. S. Noh and J. A. Schwarz, J. Catal., 1988, 114, 433–439 CrossRef CAS.
- Z. Y. Zhou, Z. Z. Huang, D. J. Chen, Q. Wang, N. Tian and S. G. Sun, Angew. Chem., Int. Ed., 2010, 49, 411–414 CrossRef CAS PubMed.
- (a) Y. H. Qin, H. H. Yang, X. S. Zhang, P. Li and C. A. Ma, Int. J. Hydrogen Energy, 2010, 35, 7667–7674 CrossRef CAS; (b) R. N. Singh, A. Singh and Anindita, Int. J. Hydrogen Energy, 2009, 34, 2052–2057 CrossRef CAS; (c) R. N. Singh, A. Singh and Anindita, Carbon, 2009, 47, 271–278 CrossRef CAS; (d) L. F. Dong, R. R. S. Gari, Z. Li, M. M. Craig and S. F. Hou, Carbon, 2010, 48, 781–787 CrossRef CAS.
- Y. C. Zhao, L. Zhan, J. N. Tian, S. L. Nie and Z. Ning, Electrochim. Acta, 2011, 56, 1967–1972 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra27882a |
| This journal is © The Royal Society of Chemistry 2016 |