
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Silver nanoparticles with different size and shape: equal cytotoxicity, but different antibacterial effects†
J.
Helmlinger‡
a,
C.
Sengstock‡
b,
C.
Groß-Heitfeld
c,
C.
Mayer
c,
T. A.
Schildhauer
b,
M.
Köller
b and
M.
Epple
*a
aInorganic Chemistry and Center for Nanointegration Duisburg-Essen (CeNIDE), Universitaetsstr. 5-7, 45117 Essen, Germany. E-mail: matthias.epple@uni-due.de
bBergmannsheil University Hospital/Surgical Research, Ruhr-University of Bochum, Buerkle-de-la-Camp-Platz 1, 44789 Bochum, Germany
cPhysical Chemistry and Center for Nanointegration Duisburg-Essen (CeNIDE), Universitaetsstr. 5-7, 45117 Essen, Germany
First published on 9th February 2016
Abstract
The influence of silver nanoparticle morphology on the dissolution kinetics in ultrapure water as well as the biological effect on eukaryotic and prokaryotic cells was examined. Silver nanoparticles with different shapes but comparable size and identical surface functionalisation were prepared, i.e. spheres (diameter 40–80 and 120–180 nm; two different samples), platelets (20–60 nm), cubes (140–180 nm), and rods (diameter 80–120 nm, length > 1000 nm). All particles were purified by ultracentrifugation and colloidally stabilized with poly(N-vinyl pyrrolidone) (PVP). Their colloidal dispersion in ultrapure water and cell culture medium was demonstrated by dynamic light scattering. Size, shape, and colloidal stability were analysed by scanning electron microscopy, atomic force microscopy, dynamic light scattering, and differential centrifugal sedimentation. The dissolution in ultrapure water was proportional to the specific surface area of the silver nanoparticles. The averaged release rate for all particle morphologies was 30 ± 13 ng s−1 m−2 in ultrapure water (T = 25 ± 1 °C; pH 4.8; oxygen saturation 93%), i.e. about 10–20 times larger than the release of silver from a macroscopic silver bar (1 oz), possibly due to the presence of surface defects in the nanoparticulate state. All particles were taken up by human mesenchymal stem cells and were cytotoxic in concentrations of >12.5 μg mL−1, but there was no significant influence of the particle shape on the cytotoxicity towards the cells. Contrary to that, the toxicity towards bacteria increased with a higher dissolution rate, suggesting that the toxic species against bacteria are dissolved silver ions.
Introduction
Metallic nanoparticles are described as materials at the boundary between atoms and the macroscopic bulk material. Due to their small size, below 100 nm in at least one dimension, their specific surface area is very large and the amount of surface atoms is higher than in the bulk material.1 Specifically, silver nanoparticles are well known for their interesting properties and are widely used, e.g. in catalysis,2,3 photonics,4–6 medical applications7,8 or even energy storage and conversion.9Concerning the biological impact of silver nanoparticles, it is well known that colloidal silver has an antimicrobial effect10,11 that can be explained by damage to the cell membrane and intracellular metabolic activity.12 Colloidal silver is therefore an interesting “inorganic” alternative to classical antibiotics, especially for the use on medical surfaces and implants.13–21
In our previous studies on the biological effects of uniform spherical silver nanoparticles (PVP-coated, diameter 80 nm) on human mesenchymal stem cells (hMSC), we have shown that cell activation occurs at elevated but non-toxic concentrations,22,23 and that adipogenic and osteogenic differentiation of hMSC were attenuated even at non-toxic concentrations.24 In addition, silver nanoparticles were taken up by hMSC and monocytes as nanoparticulate material and localized as agglomerates in the perinuclear region.23,25 Furthermore, Ag+ in ionic form is much more toxic than the same molar concentration of silver in the form of nanoparticles.26 Ag+ ions are released from silver nanoparticles when oxygen is present.27–30 We have recently shown that Ag+ ions which are responsible for the majority of the biological effects on various cells will precipitate as silver chloride in biological media.30
Because a strong correlation between the morphology of silver nanocrystals and their physical and biological properties (including their dissolution rate) can be predicted, the shape-controlled synthesis of such particles has recently received growing attention. Today, a vast amount of synthetic strategies is available.31–33 Xia et al. and others have described the structural synthesis of nanosilver in the form of cubes,34–45 platelets,46–50 rods,51,52 rings,53 and bipyramids.54 However, only little is known so far about the influence of the particle morphology on their dissolution kinetics and their toxicity in biological media.
In general, size, shape, surface charge, functionalisation, and core structure are important factors that determine the biological effects of nanoparticles, such as cellular uptake, cellular activation, as well as intercellular distribution.55–60 Sotiriou et al. have argued that smaller silver particles release silver ions faster, leading to a higher toxicity due to a higher effective silver ion concentration. They found an antibacterial effect at a silver concentration between 1 and 30 ppm, depending on the silver nanoparticle size and silver content.61
Similar results for spherical silver nanoparticles with the size of 7, 29 and 89 nm were observed by Martinez-Castanon et al.62 They found that the antibacterial effect of silver nanoparticles decreased with increasing particle size. Hong et al. reported that silver nanocubes with a size of 55 nm had the highest antibacterial effect to E. coli in comparison to spheres and wires.63 However, Actis et al. reported no antibacterial effect on S. aureus with spherical, triangular and cubic silver nanoparticles.64 Favi et al. demonstrated that gold nanospheres were more toxic than gold nanorods for fibroblasts.65 With E. coli, Pal et al. found comparable results for rod-shaped, spherical, and triangular silver nanoparticles. They suggested a relation to the atom-density of surface facets.66
Thus, the published results on different shapes of silver nanoparticles are not consistent, and the state of colloidal dispersion of the nanoparticles in cell culture media (e.g. a possible agglomeration) is not always reported in the literature. Dissolution data for different nanoparticle morphologies were not reported so far. To close this gap, we have prepared highly uniform silver nanoparticles with defined size and shape, i.e. nanospheres, nanoplatelets, nanocubes and nanorods, all with the same capping agent poly(N-vinylpyrrolidone) (PVP). All particles were purified by ultracentrifugation several times to remove free silver ions and synthesis by-products and carefully analysed by different microscopic and colloidal methods. They were colloidally stable (i.e. well dispersed) in ultrapure water as well as in cell culture medium for at least 24 h at 37 °C.
In the following, we present a comprehensive study about the correlation of the morphology of silver nanoparticles with their dissolution kinetics in ultrapure water and their toxicity towards eukaryotic cells and bacteria (prokaryotic cells).
Experimental
General notes on the preparation of silver nanoparticles
The reproducible synthesis of silver nanoparticles with well-defined shapes and sizes is highly sensitive towards a large number of synthetic parameters, e.g. the supplier and the purity of the applied reagents.33,36,39,67,68 Therefore, we describe our methods below in as much detail as possible.All chemicals were used as received without further purification. All manual operations were carried out with a glass spatula to avoid contaminations with traces of iron and other metals which may influence the crystal growth and shape.69 Glass flasks and stirring bars were cleaned with aqua regia and ultrapure water (18 MΩ cm−1) several times before they were applied.
Glucose synthesis of spherical silver nanoparticles
The synthesis of spherical silver nanoparticles by reduction of silver nitrate with glucose was carried out according to Wang et al.67,70 Briefly, 2 g glucose (≥99.5%, Sigma-Aldrich, cat. no. G7528) and 1 g poly(N-vinylpyrrolidone) (PVP; MW ≈ 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, Sigma-Aldrich, cat. no. 81420) were dissolved in 40 mL ultrapure water and stirred at 90 °C for 45 min under ambient conditions. 1 mL of a 2.72 M solution of silver nitrate (≥99.9%, Carl-Roth, cat. no. 7908.1) in water was quickly injected into the hot solution, and the temperature was kept for 60 min under continuous stirring. After cooling to room temperature, the particles were collected by ultracentrifugation (29400g, 30 min) and washed with acetone and ultrapure water several times.
000 g mol−1, Sigma-Aldrich, cat. no. 81420) were dissolved in 40 mL ultrapure water and stirred at 90 °C for 45 min under ambient conditions. 1 mL of a 2.72 M solution of silver nitrate (≥99.9%, Carl-Roth, cat. no. 7908.1) in water was quickly injected into the hot solution, and the temperature was kept for 60 min under continuous stirring. After cooling to room temperature, the particles were collected by ultracentrifugation (29400g, 30 min) and washed with acetone and ultrapure water several times.
Microwave-based synthesis of spherical silver nanoparticles
Spherical silver nanoparticles were also synthesized by a microwave-assisted polyol process.71 30 mg silver nitrate (≥99.9%, Carl-Roth) and 20 mg PVP (MW ≈ 55000 g mol−1, Sigma-Aldrich) were completely dissolved in 5 mL diethylene glycol (≥99.0%, Sigma-Aldrich) at room temperature. The solution assumed a light yellow colour, indicating the formation of small Ag seeds. The vial was then sealed with a PTFE cap and transferred to the microwave reactor (CEM Discover SP; 200 W). After stirring for 1 min at room temperature, the solution was rapidly heated to 160 °C by the microwave at the maximum possible rate. The heating took about 120 s. Temperature and stirring were maintained for 20 min. During this time, the reaction was monitored with a video camera, showing a colour change from light yellow over red and green to dark brown. To stop the reaction at a defined endpoint, the solution was quickly cooled to room temperature using compressed air in the microwave within about 120 s and then diluted in ultrapure water. The particles were collected by ultracentrifugation (66000g, 30 min) and washed with acetone and ultrapure water several times.
Microwave-based synthesis of silver nanoplatelets
Silver nanoplatelets were synthesized by microwave-assisted reduction, using a protocol described by Darmanin et al.49 Silver nitrate (≥99.9%, Carl-Roth) and ethylene glycol monoethyl ether (EEE; ≥99.9%, Sigma-Aldrich) were heated in the presence of PVP (MW ≈ 10000 g mol−1, Sigma-Aldrich). The reaction was carried out in the microwave (CEM Discover SP, 25 to 100 W). It was necessary to increase manually the microwave input power from 25 W up to 100 W at about 100 °C to achieve a reaction temperature of 140 °C after about 10 min. All other parameters remained unchanged. After the reaction was finished, the product was cooled to room temperature and diluted with acetone. The particles were collected by centrifugation (6000g, 30 min) and subsequently washed with acetone, ethanol, and ultrapure water before they were finally dispersed in argon-saturated ultrapure water.
Synthesis of silver nanocubes
The synthesis of silver nanocubes by the polyol process has been extensively described in the literature,34,35,37,38,40–44 but often turns out to be difficult to reproduce.36,39,67 Here, we have used a protocol developed by Xia et al.34 and applied some modifications to optimize the particle quality. 6 mL ethylene glycol (≥99.8%, Sigma-Aldrich) were placed in a 50 mL round bottom flask, sealed with a glass stopper and heated at 140 °C under constant stirring (1300 rpm, IKA RET basic hotplate). After 60 min, 30 μL of 0.1 M HCl in water were added. With a two-channel syringe pump (KDS-200, KD Scientific), freshly prepared solutions of silver nitrate (3 mL, 16.0 mg mL−1, Carl Roth) and PVP (3 mL, 16.2 mg mL−1, MW ≈ 55000 g mol−1, Sigma-Aldrich) in ethylene glycol were simultaneously added at a rate of 45 mL h−1. The flask was then sealed with a glass stopper. Heating and stirring were continued. Due to the poor solubility of PVP in ethylene glycol, the initial solution was placed into an ultrasonic bath before injection until all visible solid PVP had been dissolved. After 20 h, the reaction was stopped by externally cooling the flask with water to room temperature and adding 10 mL of analytical grade acetone. This lead to a yellow/ochre colloidal dispersion. The particles were collected by ultracentrifugation (29400g, 30 min). To remove silver nanowires, which typically occur as a by-product, filtration was performed with 0.45 μm cellulose syringe filters. The particles were isolated by ultracentrifugation (29400g, 30 min) and washed with acetone and ultrapure water several times. The final dispersion was stored in argon-saturated ultrapure water.
Synthesis of silver nanowires
Silver nanowires were obtained by a modified polyol process where silver chloride nanocubes were generated in situ by the reaction of silver nitrate with sodium chloride as described by Buhro et al.52 The AgCl seeds served as a heterogeneous nuclei for the growth of silver nanowires. The reaction was performed in ethylene glycol (≥99.8%, Sigma-Aldrich) in the presence of PVP (MW ≈ 55000 g mol−1, Sigma-Aldrich), with a low-concentration addition of sodium chloride (≥99.9%, VWR Chemicals) and silver nitrate (≥99.9%, Carl-Roth), exactly following the protocol described in the literature.52 After the seeding was completed, silver nanowires were produced by adding 10 mL of a 0.12 M silver nitrate solution in ethylene glycol with a syringe pump (KDS-200, KD Scientific). The particles were collected by centrifugation (666g, 30 min) and redispersed in ultrapure water with ultrasonication. The particles were washed with aqueous ammonia (2 mL 30% aqueous NH3 given to 5 mL particle dispersion) to remove excess silver chloride, centrifuged again under the same conditions and rinsed with ultrapure water several times.
Storage and analytical methods
After purification, all particles were stored in argon-saturated, ultrapure water and kept in the dark to avoid dissolution or chemical aging. All particles were colloidally stable for at least 6 months under these conditions. To determine the size and shape of the metallic particle core, 10 μL of a dispersion was dripped on a silicon wafer, dried at room temperature in air, and analysed by scanning electron microscopy (SEM) with a FEI Quanta 400 ESEM instrument in high vacuum without sputtering. Atomic force microscopy (AFM) was performed with a NanoWizard from JPK (Berlin) in the intermittent contact mode. Hydrodynamic particle diameter, zeta potential and polydispersity index (PDI) were determined by dynamic light scattering (DLS) using a Malvern Zetasizer Nano ZS. The particle size distribution and colloidal stability in cell culture medium (RPMI/FBS) after 24 h at 37 °C were also measured by DLS. Furthermore, differential centrifugal sedimentation was performed for all particles with a CPS Instruments Disc Centrifuge DC 24000 at 25 °C. Two sucrose solutions (8 wt% and 24 wt%) were used to provide a density gradient with dodecane as stabilizing agent. The calibration standard was a dispersion of poly(vinyl chloride) (PVC) particles in water. The silver concentration in the particle dispersions was analysed by atomic absorption spectroscopy (Thermo Electron Corporation, M-Series, detection limit 1 μg L−1) after dissolving 500 μL of each sample in 4.5 mL concentrated nitric acid and 5 mL ultrapure water.Dissolution in ultrapure water
The dissolution kinetics were examined by dialysis in triplicate for each kind of nanoparticles. After all silver ions were removed by ultracentrifugation and redispersion in water as described above, 4 mL silver nanoparticle dispersion (0.1 mg mL−1 Ag) were transferred in dialysis bags (Spectra/Por Biotech; cellulose ester; MWCO 100000) and immersed in 396 mL ultrapure water. The dialysis bags were rinsed with ultrapure water several times before use. Temperature, pH-value and oxygen saturation of the medium were recorded during the experiment. The dialysis was carried out under slow stirring for 696 h (29 days) in sealed polypropylene-tubes. The dissolved amount of Ag+ ions was determined by atomic absorption spectroscopy on aliquots of 9 mL taken after selected times. The aliquot volume was not replenished.
The silver bar was immersed in 500 mL of ultrapure water without stirring to avoid mechanical abrasion of the metal. After 700 h, the solution was gently mixed and a sample was taken and analysed for the silver content.
Cell culture experiments
Human mesenchymal stem cells (hMSC, 3rd to 7th passage, Lonza, Walkersville Inc., MD, USA) were cultured in RPMI1640 medium (Life Technologies, Darmstadt, Germany) containing 10% fetal calf serum (FCS, Life Technologies) and L-glutamine (0.3 g L−1, Life Technologies) using 75 cm2 flasks (Falcon, Becton Dickinson GmbH, Heidelberg, Germany). Cells were maintained at 37 °C in a humidified atmosphere of 5% CO2. Depending on the cell proliferation, the cells were sub-cultivated every 7–14 days. Adherent cells were washed with phosphate-buffered saline solution (PBS, Life Technologies) and detached from the culture flasks by addition of 0.2 mL cm−2 0.25% trypsin/0.05% ethylendiamine tetraacetic acid (EDTA, Sigma-Aldrich, Taufkirchen, Germany) for 5 min at 37 °C. Subsequently, the hMSC were collected and washed twice with RPMI1640/10% FCS. To determine the effect of silver nanoparticles on cell viability and morphology, hMSC were incubated with different concentrations of silver nanoparticles under cell culture conditions. After 24 h of incubation, the treated cells were stained with the cell-permeant dye calcein-acetoxymethylester (calcein-AM, Calbiochem, Schwalbach, Germany). For this, the nanoparticle-treated cells were washed twice with RPMI and incubated with calcein-AM (1 mM) at 37 °C for 30 min under cell culture conditions. Subsequently, the adherent cells were washed with RPMI and analysed by fluorescence microscopy (Olympus MVX10, Olympus, Hamburg, Germany). Fluorescence microphotographs were taken (CellP, Olympus), and digitally processed using AdobePhotoshop® 7.0.Confocal laser scanning microscopy (CLSM) was used to detect intracellular silver nanoparticles in hMSC after incubation. Therefore, hMSC were subconfluently grown on 2-well Lab-Tek™ glass chamber slides (Thermo Fisher Scientific, Langenselbold, Germany), and subsequently washed and exposed to 12.5 μg mL−1 silver nanoparticles for 24 h under cell culture conditions. After this incubation, the cells were incubated with 162 μM Hoechst33342 in pure RPMI1640 for 5 min at 37 °C for labelling of the nucleus. After three rinses in RPMI1640, the cells were put on glass chamber slides. Images were taken using a 40× oil immersion objective in CLSM (LSM700; Zeiss) and Zeiss 2010 software.
Bacterial culture experiments
Antimicrobial tests were performed with Staphylococcus aureus (DSMZ 1104) obtained from Life Technologies GmbH (Karlsruhe, Germany). Bacterial concentrations of overnight cultures were measured using a Densichek® (bioMérieux, Lyon, France) turbidity photometer. The calculation of bacterial counts was based on turbidity standard solutions (McFarland scale).The antimicrobial effect of silver nanoparticles with different morphologies was tested using a standard method that determines the minimum bactericidal concentration (MBC). The MBC was determined in RPMI 1640 containing 10% (v/v) fetal calf serum and L-glutamine and defined as the lowest silver concentration that completely prevented colony forming units (CFU) on the agar plate. Working silver stock solutions with different concentrations (50 μL) were added to different bacteria concentrations (102 to 105 mL−1) for inoculation. Cells were incubated in a cell culture incubator (RPMI/10% FCS) at 37 °C overnight. The minimum bactericidal concentration (MBC) was subsequently determined by plating 100 μL aliquots of the samples on blood agar platelets.
Statistical analysis
Data are expressed as mean ± SD of four independent experiments. Analysis of the data distribution was performed using the Student's t-test to analyze the significance of differences between the treated group and the control group (without silver exposure). p values of less than 0.05 were considered as statistically significant.Results and discussion
The reproducible synthesis of silver nanoparticles with defined morphologies is very challenging due to the high number of reaction parameters that must be monitored and controlled.36,39 Furthermore, if the particles shall be applied in biological systems as antibacterial agent, they must satisfy specific requirements. Size and shape should be analysed by different analytical methods, the particles must be separated from synthesis by-products that might interfere with the biological system, the colloidal stability in cell culture medium must be given at physiological temperatures over the period of the experiments, and the concentration must be high enough to cause a significant biological effect.Based on these requirements, we have chosen different syntheses from the literature and modified them if necessary. We have also taken into account that all particles are stabilized with the same ligand (PVP) to minimize a possible influence of the organic shell on the particle properties. Of course, it is impossible to prepare particles with exactly the same size, but with different shape. We have prepared two different types of spherical particles, one from a glucose synthesis72 and one from a microwave synthesis.71 The spherical particles from the microwave synthesis are about two times larger than the particles from the glucose synthesis. Fig. 1 schematically shows the size and morphology of the different particles.
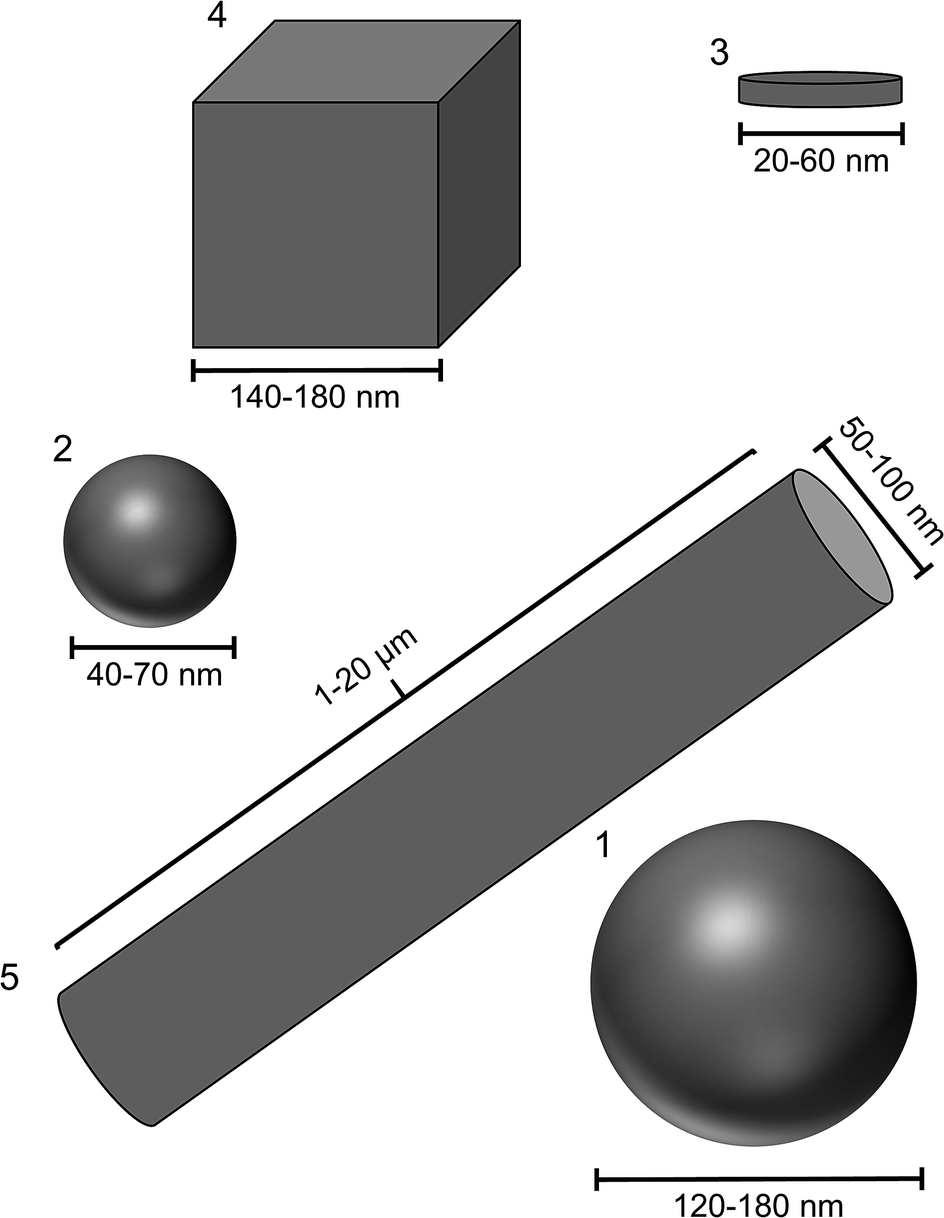 | ||
| Fig. 1 Size and shape of the different silver nanoparticles. (1) Spheres from glucose synthesis; (2) spheres from microwave synthesis; (3): platelets; (4): cubes; (5): rods (longer than shown here). | ||
All particles were highly uniform in size and shape as shown by scanning electron microscopy (Fig. 2). The dimensions of the metallic particle cores are summarized in Table 1. The base area of the platelets was either triangular or circular as described in the literature.49 The cubes had very sharp edges. A very small amount of prisms and pentagonal decahedra occurred as by-products during the synthesis. The rods were highly anisotropic with diameters in the range of 50–100 nm and lengths up to 20 μm. Although it must be acknowledged that it is impossible to prepare silver nanoparticles with 100% identical size and shape, the differences among the samples 1 to 5 (Fig. 1) are clearly larger than the variation within each sample.
 | ||
| Fig. 2 Representative SEM images of (A) silver nanospheres from glucose synthesis, (B) silver nanospheres from microwave synthesis, (C) silver nanoplatelets, (D) silver nanocubes, and (E) silver nanorods. (F) Shows the size distribution of all particles except silver nanorods in ultrapure water as obtained by differential centrifugal sedimentation. | ||
| Spheres glucose synthesis | Spheres microwave synthesis | Platelets | Cubes | Rods | |
|---|---|---|---|---|---|
| SEM | 40–70 nm | 120–180 nm | 20–60 nm | 140–180 nm | Diameter: 50–100 nm, length: 1000–20000 nm |
| DCS | 45 nm | 123 nm | 30 nm | 167 nm | — |
| DLS | 108 nm | 128 nm | 48 nm | 211 nm | 558 nm |
| AFM | Diameter: 145 nm, height: 48 nm | — | Diameter: 80 nm, height: 16 nm | — | — |
| Zeta potential by DLS | −6 mV | −3 mV | −7 mV | −11 mV | −20 mV |
Differential centrifugal sedimentation was performed with all particles. This method relies on the sedimentation of nanoparticles in a density gradient under centrifugal force. We were not able to acquire meaningful data for the silver nanorods because they were sedimenting too fast even at the lowest possible disc speed. Note that differential centrifugal sedimentation tends to underestimate the particle diameters because the density of the pure metal is used in the calculation, while the effective density is lower due to the organic ligand shell. On the other hand, the polymer- and hydration layers are contributing to the particle diameters. Furthermore, the particles are assumed as spherical, leading to deviations for the platelets and cubes. Nevertheless, the results (Table 1) reasonably agree with the electron microscopic data.
The size distribution in solution and the colloidal stability were determined by dynamic light scattering (DLS), and the zeta potential in ultrapure water was measured. The results are given in Table 1 and Fig. S1 (ESI†). All particles were colloidally stable in ultrapure water as well as in cell culture medium (RPMI/FBS) over the observation period of 24 h at 37 °C. In cell culture medium, the particle diameters slightly increased, probably due to the formation of a protein corona.45 Similar to differential centrifugal sedimentation, the particles are assumed as spherical, leading to deviations for silver nanorods. All other diameters agreed well with the results obtained by SEM. The zeta potential was negative in all cases, i.e. the particles were electrosterically stabilized.
As it is difficult to determine the height of silver nanoplatelets by scanning electron microscopy, we have performed atomic force microscopy on platelets and spheres from the glucose synthesis. Fig. 3 shows a typical line scan analysis as well as a magnified image for both kinds of particles. We manually selected ten particles from each sample and determined their average width and height. The mean value for the thickness of the platelets was 16 nm and agreed to the value of 10–15 nm as reported by Darmanin et al.49 We also calculated the aspect ratio for both kind of particles. The ratio was 5:1 for the platelets and 3:1 for the spheres.
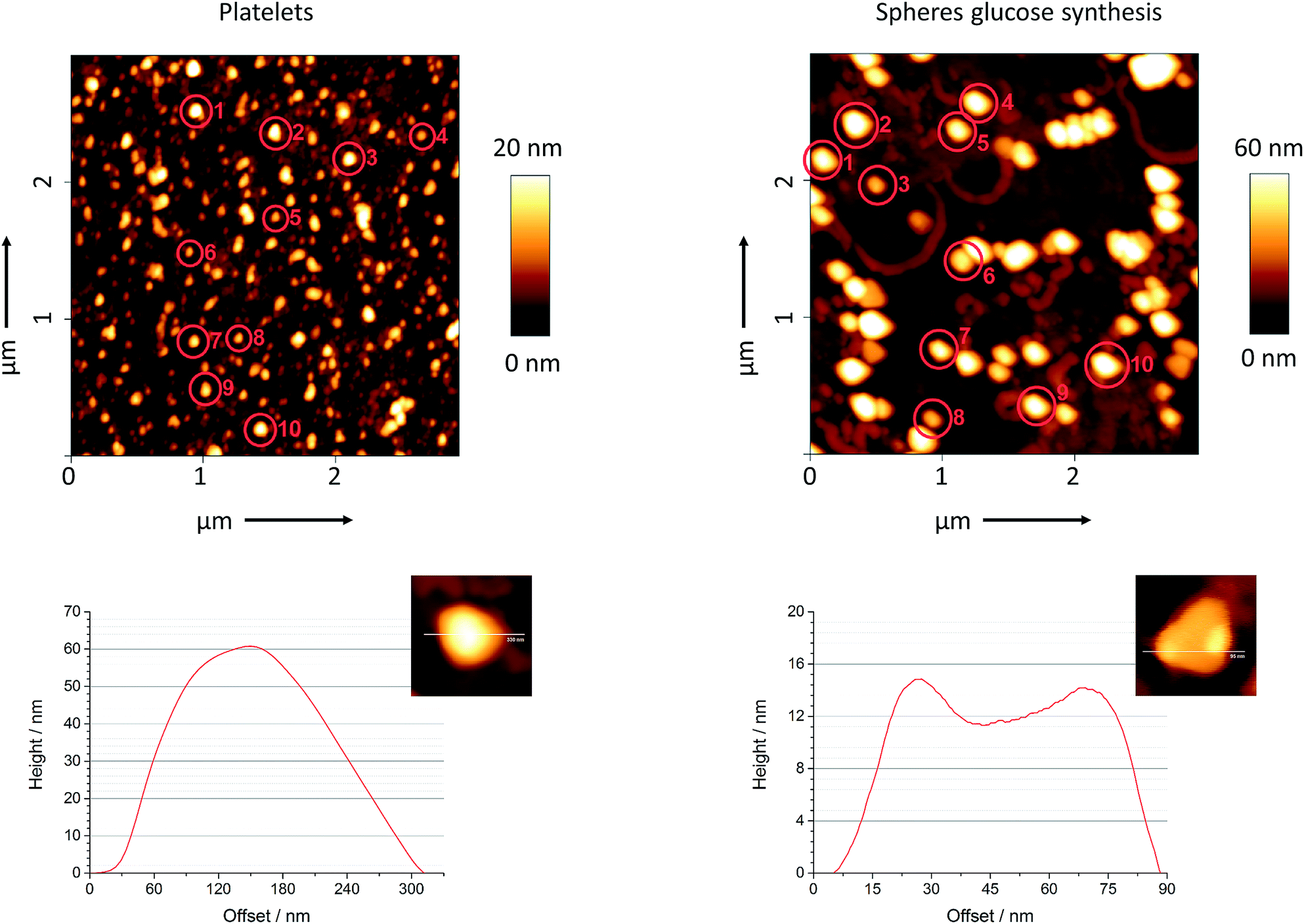 | ||
| Fig. 3 Topology of silver nanoplatelets (left) and silver nanospheres from glucose synthesis (right). Ten individual particles were selected from each sample and their diameter and height were manually measured by atomic force microscopy. | ||
The influence of the particle morphology on the dissolution kinetics of silver nanoparticles was examined by dialysis. It was shown that silver nanoparticles dissolve in water under the formation of Ag+ ions when oxygen is present.27,29,30,73–78 The released Ag+ ions are small enough to pass a cellulose-ester membrane and can be detected by AAS, while the nanoparticles are retained inside the dialysis bag.27,30
Fig. 4 shows the dissolved amount of Ag+ from nanoparticles as function of time. We chose ultrapure water as the dissolution medium (T = 25 ± 1 °C; pH = 4.8; oxygen saturation = 93%), which is of course a simplified model system. Diendorf et al. have shown that in biological media, complexation and precipitation by biomolecules and salts must be taken into account because they significantly influence the dissolution kinetics.30
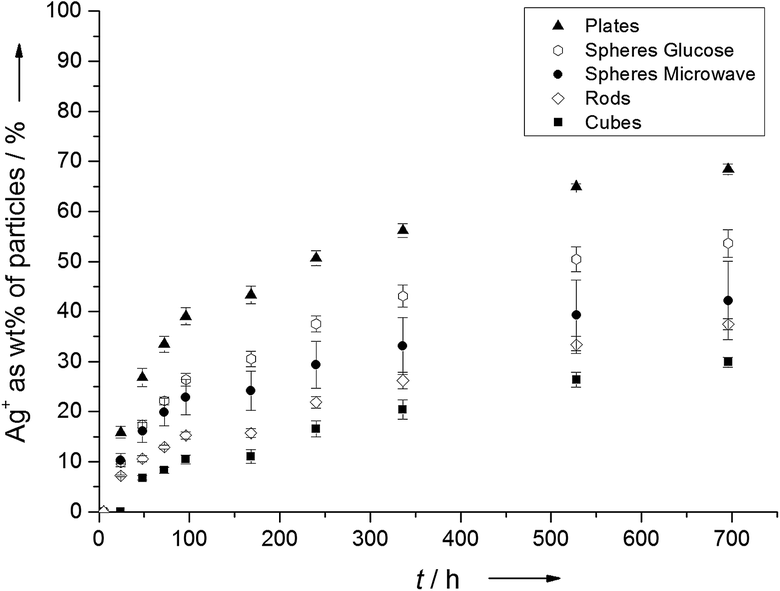 | ||
| Fig. 4 Experimentally determined dissolution kinetics of silver nanoparticles with different morphologies. | ||
It is obvious from Fig. 4 that the particle morphology has a significant effect on the release of Ag+ ions. After 696 hours (29 days), 68 ± 1 wt% Ag of the platelets were dissolved as Ag+, followed by spheres from the glucose synthesis (54 ± 3 wt% Ag), spheres from the microwave synthesis (42 ± 8 wt% Ag), rods (37 ± 1 wt% Ag) and finally cubes (30 ± 1 wt% Ag). As expected, the amount of dissolved silver from the glucose particles matched the value of ∼53% wt% Ag which was obtained by Kittler et al. for the same particles under the same conditions.27
We have estimated the specific surface area for all particles by simplified geometric calculations and correlated the results with the experimentally determined dissolution kinetics. For this, we have calculated the volume and surface for the particles, assuming ideal spheres and cubes. Platelets and rods are assumed as cylindrical with a circular base. For particle radii, heights and edge lengths we have chosen reasonable average values for the metallic particle core from experimentally obtained scanning electron micrographs (Table 2).
| Spheres glucose synthesis | Spheres microwave synthesis | Platelets | Cubes | Rods | Silver bar (1 oz) | |
|---|---|---|---|---|---|---|
| Radius/nm | 30 | 75 | 20 | — | 50 | — |
| Height/nm | — | — | 15 | — | 4000 | 2.2 × 106 (2.2 mm) |
| Edge length/nm | — | — | — | 160 | — | 5 × 107 (50 mm), 2.8 × 107 (28 mm) |
| Volume per particle/nm3 | 1.13 × 105 | 1.77 × 106 | 1.88 × 104 | 4.10 × 106 | 3.14 × 107 | 2.98 × 1021 |
| Surface area per particle/nm2 | 1.13 × 104 | 7.07 × 104 | 4.40 × 103 | 1.54 × 105 | 1.27 × 106 | 3.18 × 1015 |
| Surface area/volume per particle/nm−1 | 0.100 | 0.040 | 0.234 | 0.038 | 0.040 | 1.07 × 10−6 |
| Particle concentration/mL−1 | 8.43 × 1010 | 5.39 × 109 | 5.06 × 1011 | 2.33 × 109 | 3.03 × 108 | — |
| Total particle surface/m2 mL−1 | 9.54 × 10−4 | 3.81 × 10−4 | 2.22 × 10−3 | 3.58 × 10−4 | 3.86 × 10−4 | — |
| Dissolution of Ag per time/μg mL−1 s−1 (average after 696 h) | 2.14 × 10−5 | 1.68 × 10−5 | 2.73 × 10−5 | 1.19 × 10−5 | 1.49 × 10−5 | 6.24 × 10−6 |
| Dissolution rate of Ag per time and surface area/ng s−1 m−2 | 22 | 44 | 12 | 33 | 39 | 2 |
The trend of the dissolution rate was as follows (Table 2):
| platelets > spheres glucose synthesis > spheres microwave synthesis ≈ rods > cubes |
The specific surface area follows the same trend. When compared to the experimentally determined dissolution rates (Fig. 4), these results indicate a strong correlation between the specific surface area and the dissolution kinetics for silver nanoparticles. Particles with a higher specific surface area dissolve faster than those with a smaller specific surface area, leading to the assumption that the oxidation of Ag to Ag+ on the particle surface is the rate-determining step of the dissolution.
If the silver release rate is normalized to the particle surface, values between 12 and 44 ng s−1 m−2 are obtained. On average, the silver dissolution rate is 30 ± 13 ng s−1 m−2. Here, differences in the silver release rate from different crystallographic faces are neglected. Remarkably, the dissolution rate of a silver bar (1 oz) immersed in pure water for 696 h was about one order of magnitude smaller (2 ng s−1 m−2 compared to the nanoparticles). This can tentatively ascribed to defects in the surface of nanoparticles compared to a macroscopic metallic alloy.
The biological effect of the different silver nanoparticle shapes on prokaryotic and eukaryotic cells was analysed. To ensure comparable conditions, we have used RPMI 1640 with 10% (v/v) FCS as culture medium in both cases. This medium is typically used for the cultivation of mammalian cells. It represents a good approximation of in vivo conditions where the antibacterial action is desired. In addition, we have already shown that PVP-stabilized silver nanoparticles remain well dispersed in this medium.79
Human mesenchymal stem cells (hMSC) as typical tissue-like cells were cultured in the presence of 12.5 μg mL−1 silver nanoparticles at 37 °C for 24 h under cell culture conditions. The uptake of the silver nanoparticles was analysed by CLSM. Nanoplatelets were visible in the perinuclear region as agglomerates (white arrow) close to the cell nucleus but not within the cell nucleus (Fig. 5A). The cell nucleus was labelled by using Hoechst33342 (blue fluorescence). Similar results were observed for the nanoparticles of other shapes (see ESI Fig. S2†). In previous studies we have shown by combined FIB/SEM and EDX that hMSC are able to internalize spherical silver nanoparticles.25
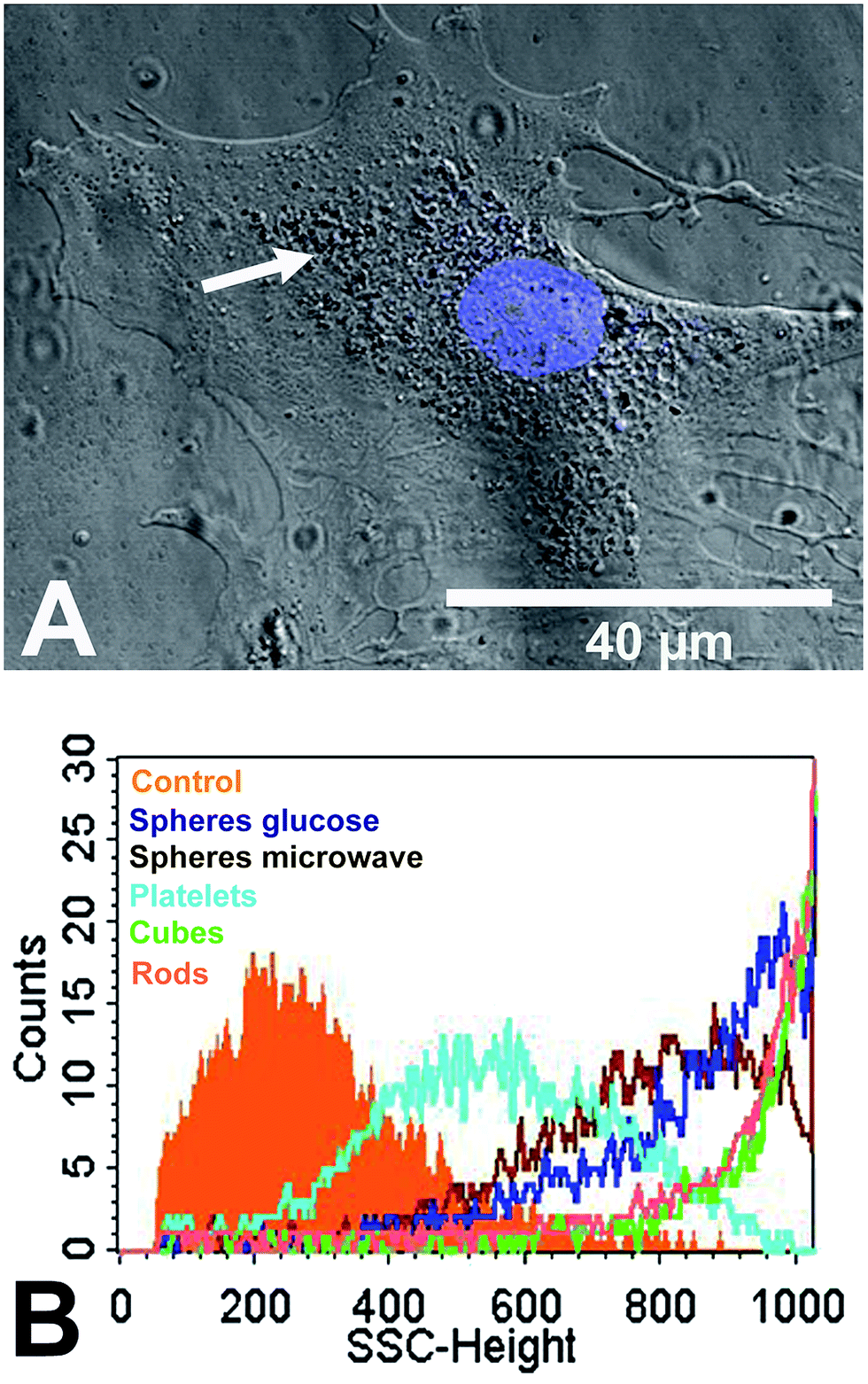 | ||
| Fig. 5 Intracellular occurrence of silver nanoparticles agglomerates analysed by phase-contrast microscopy and flow cytometric side scatter analysis. (A) Representative phase-contrast micrograph of hMSC treated with 12.5 μg mL−1 silver nanoplatelets at 37 °C for 24 h under cell culture conditions. The white arrow denotes perinuclear accumulation of nanoparticles. The blue fluorescence of Hoechst33342 was used to stain the cell nucleus. (B) A representative flow cytometric histogram of the side scatter intensity signal for hMSC treated without (control) or with different silver nanoparticles morphologies. | ||
The uptake of silver was additionally analysed by flow cytometry using the side-scatter signal (Fig. 5B). Flow cytometry was also used by Stringer et al. and Suzuki et al. after the uptake of nanoparticles into cells.80,81 The scattering intensity is increased by the increased light scattering from the dense ingested particles. In our study, the size of the internalized nano- or agglomerated particles (Table 1) clearly correlated with the side-scatter intensity signal (SSC-signal; Fig. 5B). The SSC-signal is correlated with the intensity of the light scattered by a particle, and this is increasing with particle or agglomerate size. Therefore, the increased SSC-signal of the nanoparticle-treated cells reflects the size of the internalized particles/agglomerates rather than an uptake rate or the particle number.
The viability of the nanoparticle-treated cells was analysed by calcein-AM staining and fluorescence microscopy. After incubation of hMSC with silver nanoparticles an increasing, dose-dependent cytotoxic effect occurred (Fig. 6). There was a statistically significant effect on cell viability in the presence of >12.5 μg mL−1 silver nanoparticles with different morphologies compared to cells treated without silver (0 μg mL−1). For spherical silver nanoparticles, similar results were reported by our group.24,26 It is remarkable that there was no silver nanoparticle shape-dependent effect on hMSC under these cell culture conditions (Fig. 6).
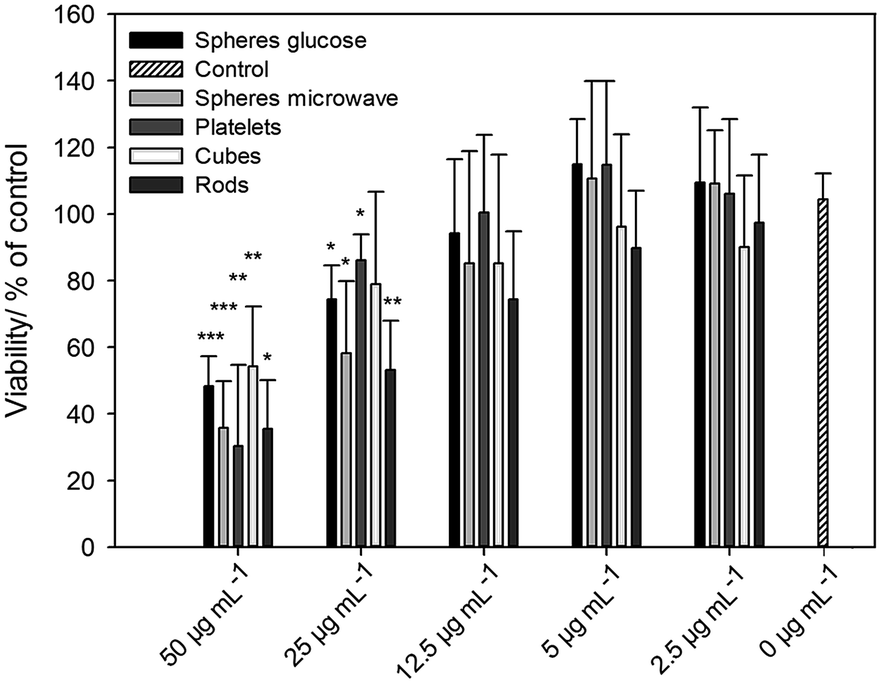 | ||
| Fig. 6 Effects of silver nanoparticles with different morphologies on the viability of cells as analysed by calcein-AM staining and digital image processing (phase analysis). The hMSC were treated with different concentrations of silver nanoparticles (μg mL−1) for 24 h under cell culture conditions. The quantitative data are expressed as the mean ± SD of four independent experiments, given as a percentage of the control (cells cultured without silver, 0 μg mL−1). An asterisk (*) indicates significant differences in comparison to the control without silver (*p < 0.05, **p < 0.005, ***p < 0.001). | ||
In addition to the biological effect of silver nanoparticles to mammalian cells, the antimicrobial effect of silver nanoparticles towards S. aureus was studied by the determination of the minimal bactericidal concentration (MBC). As represented in Fig. 7, silver nanoparticles (25 μg mL−1) showed an antimicrobial effect on S. aureus and in addition an increase in the MBC when the bacterial number was increased (see different quadrants in Fig. 7). Comparable results have been described in the literature for β-lactam antibiotics.82 This effect is well known as the inoculum effect.26,83 Interestingly, in addition to this effect, a particle shape-dependent effect on antibacterial activity was observed.
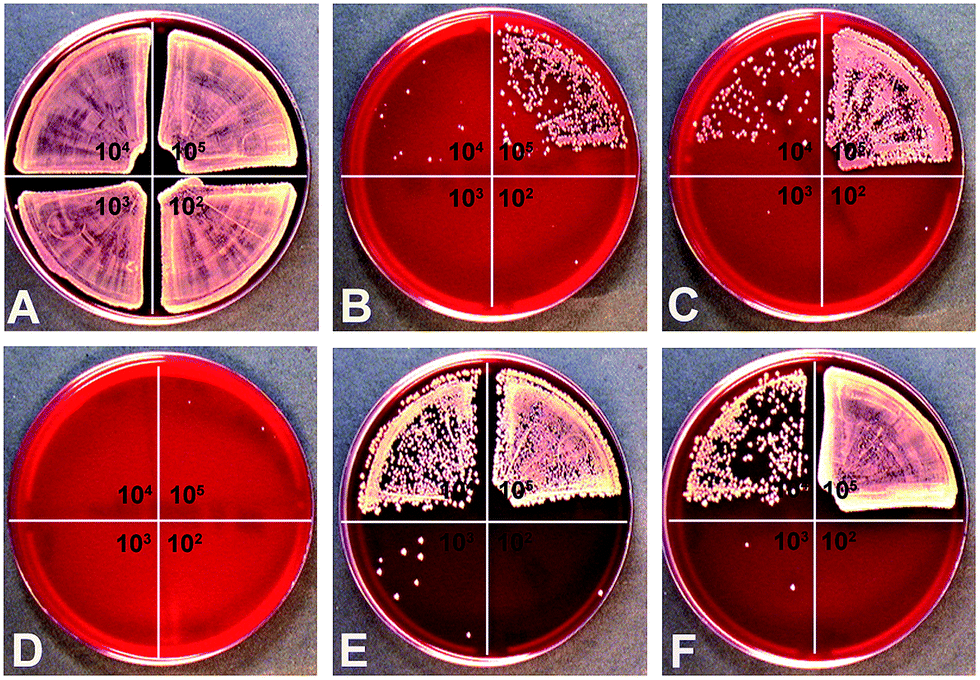 | ||
| Fig. 7 Effects of silver nanoparticles with different shape on the viability of S. aureus. Representative images of S. aureus colonies on blood agar platelets. S. aureus with different bacteria concentrations (top right quadrant 1 × 105; lower right quadrant 1 × 102; top left quadrant 1 × 104; lower left quadrant 1 × 103) were treated without (A) or with 25 μg mL−1 silver nanoparticles with different particles shapes ((B): sphere from glucose synthesis; (C): spheres from microwave synthesis; (D): platelets; (E): cubes and (F): rods) under cell culture conditions. After 24 h the bacteria were plated (100 μL) on blood agar platelets and incubated for another 24 h at 37 °C to the determine the MBC. | ||
Silver nanoplatelets showed the highest antibacterial effect, followed by spheres from the glucose synthesis, spheres from the microwave synthesis, rods, and finally cubes (Table 3). This toxic effect towards S. aureus correlates to the silver ion release (Fig. 4).
| S. aureus (N) | 1 × 105 | 1 × 104 | 1 × 103 | 1 × 102 |
| Spheres glucose synthesis | >50 | ≥50 | ≥25 | ≥12.5–25 |
| Spheres microwave synthesis | >50 | ≥50 | ≥25 | ≥25 |
| Platelets | ≥50 | ≥25 | ≥25 | ≥25 |
| Cubes | >50 | >50 | ≥50 | ≥25–50 |
| Rods | >50 | ≥50 | ≥25 | ≥25 |
We have shown that particles with a higher specific surface area such as platelets dissolve faster than particles with a smaller specific surface area such as cubes. This increased silver ion release for particles with higher specific surface area was obviously the reason for the observed increased antimicrobial effect. However, this effect was observed only for bacteria. The antibacterial effect was related to the shape of the silver nanoparticles at same total silver concentration, but this was not observed for eukaryotic cells.
We therefore suggest that the antibacterial mechanism is only related to the silver ion release because bacteria are probably unable to ingest the used silver nanoparticles in contrast to hMSC.25 Wang et al. demonstrated that 16 nm gold nanospheres adhered well to the surface of Salmonella typhimurium, but were unable to get into the bacteria.84 In another study, Butler et al. demonstrated by transmission electron microscopy that silver nanoparticles with a size of 10 nm or larger were not internalized into S. typhimurium or E. coli.85 In general, there are only few studies in the literature on the uptake of nanoparticles in bacteria. Kumar et al. have shown by flow cytometry that nanoparticles of zinc oxide (ZnO) and titanium dioxide (TiO2) were taken up by E. coli.86 However, the authors did not distinguish between internalized and adherent nanoparticles. Butler et al. have shown by transmission electron microscopy and energy dispersive X-ray spectroscopy that TiO2 nanoparticles were associated with bacteria (surrounding them almost completely), but that they were not internalized.87 In contrast to the interaction with bacteria, we have shown earlier that hMSC are able to internalize spherical silver nanoparticles, and that most of the internalized silver nanoparticles are present in the endo-lysosomal cell compartment of hMSC.25,88,89 Compared to the observed shape-dependent antibacterial effect, there was no difference in the cytotoxic effect towards hMSC.
Conclusions
In the presence of oxygen, silver nanoparticles of all shapes dissolve in pure water. The dissolution kinetics were correlated to the estimated specific surface area of the particles, where particles with a higher specific surface area dissolve faster than particles with a smaller one. By confocal laser scanning microscopy, it was shown that all particles were taken up by endocytosis of human mesenchymal stem cells. It can be assumed that they start to dissolve in the endolysosomes. This particle uptake was not shape-dependent, probably due to the short uptake time (several hours) where the dissolution rate is similar for all kinds of silver nanoparticles. A toxic effect to these cells was observed at concentrations > 12.5 μg mL−1, but no shape dependence was found. Contrary to that, the toxicity towards bacteria corresponded very well to the dissolution kinetics and therefore to the particle morphology. Particles with a higher specific surface area were more toxic for bacteria than particles with smaller specific surface areas. This suggests that Ag+ is the predominant toxic species towards bacteria. The difference in the dissolution rate may be exploited to apply silver nanoparticles with a relatively higher bacterial effect with a lower cytotoxic effect towards tissue.Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (DFG) for funding in the framework of the priority program 1313. We thank Dr Joerg Diendorf for experimental assistance with the dissolution of the silver bar.Notes and references
- G. Schmid, Nanoparticles. From Theory to Application, Wiley-VCH, Weinheim, 2004 Search PubMed.
- X. Y. Dong, Z. W. Gao, K. F. Yang, W. Q. Zhang and L. W. Xu, Catal. Sci. Technol., 2015, 5, 2554–2574 CAS.
- Y. Gao, P. Jiang, L. Song, L. Liu, X. Yan, Z. Zhou, D. Liu, J. Wang, H. Yuan, Z. Zhang, X. Zhao, X. Dou, W. Zhou, G. Wang and S. Xie, J. Phys. D: Appl. Phys., 2005, 38, 1061–1067 CrossRef CAS.
- H. Choi, S. J. Ko, Y. Choi, P. Joo, T. Kim, B. R. Lee, J. W. Jung, H. J. Choi, M. Cha, J. R. Jeong, I. W. Hwang, M. H. Song, B. S. Kim and J. Y. Kim, Nat. Photonics, 2013, 7, 732–738 CrossRef CAS.
- M. S. Hu, H. L. Chen, C. H. Shen, L. S. Hong, B. R. Huang, K. H. Chen and L. C. Chen, Nat. Mater., 2006, 5, 102–106 CrossRef CAS PubMed.
- M. Kauranen and A. V. Zayats, Nat. Photonics, 2012, 6, 737–748 CrossRef CAS.
- X. Chen and H. J. Schluesener, Toxicol. Lett., 2008, 176, 1–12 CrossRef CAS PubMed.
- P. D. Howes, R. Chandrawati and M. M. Stevens, Science, 2014, 346, 1247390 CrossRef PubMed.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- S. Chernousova and M. Epple, Angew. Chem., Int. Ed., 2013, 52, 1636–1653 CrossRef CAS PubMed.
- B. Nowack, H. F. Krug and M. Height, Environ. Sci. Technol., 2011, 45, 1177–1183 CrossRef CAS PubMed.
- W. K. Jung, H. C. Koo, K. W. Kim, S. Shin, S. H. Kim and Y. H. Park, Appl. Environ. Microbiol., 2008, 74, 2171–2178 CrossRef CAS PubMed.
- K. Markowska, A. M. Grudniak and K. I. Wolska, Acta Biochim. Pol., 2013, 60, 523–530 Search PubMed.
- W. Zimmerli, J. Intern. Med., 2014, 276, 111–119 CrossRef CAS PubMed.
- M. Beattie and J. Taylor, J Clin. Nurs., 2011, 20, 2098–2108 CrossRef PubMed.
- D. M. Eby, H. R. Luckarift and G. R. Johnson, ACS Appl. Mater. Interfaces, 2009, 1, 1553–1560 CAS.
- F. Furno, K. S. Morley, B. Wong, B. L. Sharp, P. L. Arnold, S. M. Howdle, R. Bayston, P. D. Brown, P. D. Winship and H. J. Reid, J. Antimicrob. Chemother., 2004, 54, 1019–1024 CrossRef CAS PubMed.
- M. L. W. Knetsch and L. H. Koole, Polymers, 2011, 3, 340–366 CrossRef CAS.
- D. R. Monteiro, L. F. Gorup, A. S. Takamiya, A. C. Ruvollo-Filho, E. R. Camargo and D. B. Barbosa, Int. J. Antimicrob. Agents, 2009, 34, 103–110 CrossRef CAS PubMed.
- K. N. J. Stevens, S. Croes, R. S. Boersma, E. E. Stobberingh, C. van der Marel, F. H. van der Veen, M. L. W. Knetsch and L. H. Koole, Biomaterials, 2011, 32, 1264–1269 CrossRef CAS PubMed.
- K. Vasilev, V. Sah, K. Anselme, C. Ndi, M. Mateescu, B. Dollmann, P. Martinek, H. Ys, L. Ploux and H. J. Griesser, Nano Lett., 2009, 10, 202–207 CrossRef PubMed.
- C. Greulich, S. Kittler, M. Epple, G. Muhr and M. Köller, Langenbecks Arch. Surg., 2009, 394, 495–502 CrossRef CAS PubMed.
- C. Greulich, J. Diendorf, J. Geßmann, T. Simon, T. Habijan, G. Eggeler, T. A. Schildhauer, M. Epple and M. Köller, Acta Biomater., 2011, 7, 3505–3514 CrossRef CAS PubMed.
- C. Sengstock, J. Diendorf, M. Epple, T. A. Schildhauer and M. Köller, Beilstein J. Nanotechnol., 2014, 5, 2058–2069 CrossRef CAS PubMed.
- C. Greulich, J. Diendorf, T. Simon, G. Eggeler, M. Epple and M. Köller, Acta Biomater., 2011, 7, 347–354 CrossRef CAS PubMed.
- C. Greulich, D. Braun, A. Peetsch, J. Diendorf, B. Siebers, M. Epple and M. Koller, RSC Adv., 2012, 2, 6981–6987 RSC.
- S. Kittler, C. Greulich, J. Diendorf, M. Köller and M. Epple, Chem. Mater., 2010, 22, 4548–4554 CrossRef CAS.
- J. Liu, D. A. Sonshine, S. Shervani and R. H. Hurt, ACS Nano, 2010, 4, 6903–6913 CrossRef CAS PubMed.
- L. Liu and R. H. Hurt, Environ. Sci. Technol., 2010, 44, 2169–2175 CrossRef PubMed.
- K. Loza, J. Diendorf, C. Greulich, L. Ruiz-Gonzales, J. M. Gonzalez-Calbet, M. Vallet-Regi, M. Koeller and M. Epple, J. Mater. Chem. B, 2014, 2, 1634–1643 RSC.
- Y. Xia, X. Xia and H. C. Peng, J. Am. Chem. Soc., 2015, 137, 7947–7966 CrossRef CAS PubMed.
- Y. Xia, X. Xia, Y. Wang and S. Xie, MRS Bull., 2013, 38, 335–344 CrossRef CAS.
- Y. N. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- S. H. Im, Y. T. Lee, B. Wiley and Y. Xia, Angew. Chem., Int. Ed., 2005, 44, 2154–2157 CrossRef CAS PubMed.
- H. Mehenni, L. Sinatra, R. Mahfouz, K. Katsiev and O. M. Bakr, RSC Adv., 2013, 3, 22397–22403 RSC.
- E. V. Panfilova, B. N. Khlebtsov, A. M. Burov and N. G. Khlebtsov, Colloid J., 2012, 74, 99–109 CrossRef CAS.
- M. Rycenga, J. M. McLellan and Y. Xia, Adv. Mater., 2008, 20, 2416–2420 CrossRef CAS.
- A. R. Siekkinen, J. M. McLellan, J. Chen and Y. Xia, Chem. Phys. Lett., 2006, 432, 491–496 CrossRef CAS PubMed.
- S. E. Skrabalak, L. Au, X. D. Li and Y. N. Xia, Nat. Protoc., 2007, 2, 2182–2190 CrossRef CAS PubMed.
- Y. G. Sun and Y. N. Xia, Science, 2002, 298, 2176–2179 CrossRef CAS PubMed.
- Y. Wang, Y. Q. Zheng, C. Z. Huang and Y. N. Xia, J. Am. Chem. Soc., 2013, 135, 1941–1951 CrossRef CAS PubMed.
- D. Yu and V. W. W. Yam, J. Am. Chem. Soc., 2004, 126, 13200–13201 CrossRef CAS PubMed.
- D. B. Yu and V. W. W. Yam, J. Phys. Chem. B, 2005, 109, 5497–5503 CrossRef CAS PubMed.
- Q. Zhang, W. Li, C. Moran, J. Zeng, J. Chen, L. Wen and Y. N. Xia, J. Am. Chem. Soc., 2010, 132, 11372–11378 CrossRef CAS PubMed.
- T. Miclaus, V. E. Bochenkov, R. Ogaki, K. A. Howard and D. S. Sutherland, Nano Lett., 2014, 14, 2086–2093 CrossRef CAS PubMed.
- S. Chen, Z. Fan and D. L. Carroll, J. Phys. Chem. B, 2002, 106, 10777–10781 CrossRef CAS.
- E. Hao, K. L. Kelly, J. T. Hupp and G. C. Schatz, J. Am. Chem. Soc., 2002, 124, 15182–15183 CrossRef CAS PubMed.
- Y. J. Xiong, A. R. Siekkinen, J. G. Wang, Y. D. Yin, M. J. Kim and Y. N. Xia, J. Mater. Chem., 2007, 17, 2600–2602 RSC.
- T. Darmanin, P. Nativo, D. Gilliland, G. Ceccone, C. Pascual, B. De Berardis, F. Guittard and F. Rossi, Colloids Surf., A, 2012, 395, 145–151 CrossRef CAS.
- A. Knauer and J. M. Köhler, Chem. Ing. Tech., 2013, 85, 467–475 CrossRef CAS.
- Y. Sun, B. Mayers, T. Herricks and Y. Xia, Nano Lett., 2003, 3, 955–960 CrossRef CAS.
- W. M. Schuette and W. E. Buhro, ACS Nano, 2013, 7, 3844–3853 CrossRef CAS PubMed.
- A. A. Zinchenko, K. Yoshikawa and D. Baigl, Adv. Mater., 2005, 17, 2820–2823 CrossRef CAS.
- B. J. Wiley, Y. Xiong, Z. Y. Li, Y. Yadong Yin and Y. Xia, Nano Lett., 2006, 6, 765–768 CrossRef CAS PubMed.
- O. Harush-Frenkel, N. Debotton, S. Benita and Y. Altschuler, Biochem. Biophys. Res. Commun., 2007, 353, 26–32 CrossRef CAS PubMed.
- K. Cha, H. W. Hong, Y. G. Choi, M. J. Lee, J. H. Park, H. K. Chae, G. Ryu and H. Myung, Biotechnol. Lett., 2008, 30, 1893–1899 CrossRef CAS PubMed.
- Y. C. Chung, I. H. Chen and C. J. Chen, Biomaterials, 2008, 29, 1807–1816 CrossRef CAS PubMed.
- L. Kastl, D. Sasse, V. Wulf, R. Hartmann, J. Mircheski, C. Ranke, S. Carregal-Romero, J. A. Martínez-López, R. Fernández-Chacón, W. J. Parak, H. P. Elsasser and P. Rivera-Gil, ACS Nano, 2013, 7, 6605–6618 CrossRef CAS PubMed.
- L. Shang, K. Nienhaus, X. Jiang, L. Yang, K. Landfester, V. Mailänder, T. Simmet and G. U. Nienhaus, Beilstein J. Nanotechnol., 2014, 5, 2388–2397 CrossRef CAS PubMed.
- D. A. Kuhn, D. Vanhecke, B. Michen, F. Blank, P. Gehr, A. Petri-Fink and B. Rothen-Rutishauser, Beilstein J. Nanotechnol., 2014, 5, 1625–1636 CrossRef CAS PubMed.
- G. A. Sotiriou and S. E. Pratsinis, Curr. Opin. Chem. Eng., 2011, 1, 3–10 CrossRef CAS PubMed.
- G. A. Martinez-Castanon, N. Nino-Martinez, F. Martinez-Gutierrez, J. R. Martinez-Mendoza and F. Ruiz, J. Nanopart. Res., 2008, 10, 1343–1348 CrossRef CAS.
- X. Hong, J. Wen, X. Xiong and Y. Hu, Environ. Sci. Pollut. Res., 2015, 1–9, DOI:10.1007/s11356-015-5668-z.
- L. Actis, A. Srinivasan, J. L. Lopez-Ribot, A. K. Ramasubramanian and J. L. Ong, J. Mater. Sci.: Mater. Med., 2015, 26, 1–7 CrossRef CAS PubMed.
- P. M. Favi, M. M. Valencia, P. R. Elliott, A. Restrepo, M. Gao, H. Huang, J. J. Pavon and T. J. Webster, J. Biomed. Mater. Res., Part A, 2015, 103, 3940–3955 CrossRef CAS PubMed.
- S. Pal, Y. K. Tak and J. M. Song, Appl. Environ. Microbiol., 2007, 73, 1712–1720 CrossRef CAS PubMed.
- S. Ahlberg, A. Antonopulos, J. Diendorf, R. Dringen, M. Epple, R. Flöck, W. Goedecke, C. Graf, N. Haberl, J. Helmlinger, F. Herzog, F. Heuer, S. Hirn, C. Johannes, S. Kittler, M. Köller, K. Korn, W. G. Kreyling, F. Krombach, J. Lademann, K. Loza, E. M. Luther, M. Malissek, M. C. Meinke, D. Nordmeyer, A. Pailliart, J. Raabe, F. Rancan, B. Rothen-Rutishauser, E. Rühl, C. Schleh, A. Seibel, C. Sengstock, L. Treuel, A. Vogt, K. Weber and R. Zellner, Beilstein J. Nanotechnol., 2014, 5, 1944–1965 CrossRef PubMed.
- J. Zeng, Y. Zheng, M. Rycenga, J. Tao, Z. Y. Li, Q. Zhang, Y. Zhu and Y. N. Xia, J. Am. Chem. Soc., 2010, 132, 8552–8553 CrossRef CAS PubMed.
- B. Wiley, Y. Sun and Y. Xia, Langmuir, 2005, 21, 8077–8080 CrossRef CAS PubMed.
- H. S. Wang, X. L. Qiao, J. G. Chen and S. Y. Ding, Colloids Surf., A, 2005, 256, 111–115 CrossRef CAS.
- J. Helmlinger, M. Heise, M. Heggen, M. Ruck and M. Epple, RSC Adv., 2015, 5, 92144–92150 RSC.
- S. Banerjee, K. Loza, W. Meyer-Zaika, O. Prymak and M. Epple, Chem. Mater., 2014, 26, 951–957 CrossRef CAS.
- C. Batchelor-McAuley, K. Tschulik, C. C. M. Neumann, E. Laborda and R. G. Compton, Int. J. Electrochem. Sci., 2014, 9, 1132–1138 Search PubMed.
- R. D. Kent and P. J. Vikesland, Environ. Sci. Technol., 2012, 46, 6977–6984 CrossRef CAS PubMed.
- J. M. Zook, S. E. Long, D. Cleveland, C. L. A. Geronimo and R. I. MacCuspie, Anal. Bioanal. Chem., 2011, 401, 1993–2002 CrossRef CAS PubMed.
- C. Levard, B. C. Reinsch, F. M. Michel, C. Oumahi, G. V. Lowry and G. E. Brown, Environ. Sci. Technol., 2011, 45, 5260–5266 CrossRef CAS PubMed.
- C. M. Ho, C. K. Wong, S. K. W. Yau, C. N. Lok and C. M. Che, Chem.–Asian J., 2011, 6, 2506–2511 CrossRef CAS PubMed.
- X. Li, J. J. Lenhart and H. W. Walker, Langmuir, 2010, 26, 16690–16698 CrossRef CAS PubMed.
- S. Kittler, C. Greulich, J. S. Gebauer, J. Diendorf, L. Treuel, L. Ruiz, J. M. Gonzalez-Calbet, M. Vallet-Regi, R. Zellner, M. Köller and M. Epple, J. Mater. Chem., 2010, 20, 512–518 RSC.
- B. Stringer, A. Imrich and L. Kobzik, Cytometry, 1995, 20, 23–32 CrossRef CAS PubMed.
- H. Suzuki, T. Toyooka and Y. Ibuki, Environ. Sci. Technol., 2007, 41, 3018–3024 CrossRef CAS PubMed.
- I. Brook, Clin. Infect. Dis., 1989, 11, 361–368 CrossRef CAS.
- K. I. Udekwu, N. Parrish, P. Ankomah, F. Baquero and B. R. Levin, J. Antimicrob. Chemother., 2009, 63, 745–757 CrossRef CAS PubMed.
- S. Wang, R. Lawson, P. C. Ray and H. Yu, Toxicol. Ind. Health, 2011, 27, 547–554 CrossRef CAS PubMed.
- K. S. Butler, D. J. Peeler, B. J. Casey, B. J. Dair and R. K. Elespuru, Mutagenesis, 2015, 30, 577–591 CrossRef PubMed.
- A. Kumar, A. K. Pandey, S. S. Singh, R. Shanker and A. Dhawan, Cytometry, Part A, 2011, 79, 707–712 Search PubMed.
- K. S. Butler, B. J. Casey, G. V. Garborcauskas, B. J. Dair and R. K. Elespuru, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2014, 768, 14–22 CrossRef CAS PubMed.
- Y. Hu, K. Cai, Z. Luo, R. Zhang, L. Yang, L. Deng and K. D. Jandt, Biomaterials, 2009, 30, 3626–3635 CrossRef CAS PubMed.
- H. Y. Nam, S. M. Kwon, H. Chung, S. Y. Lee, S. H. Kwon, H. Jeon, Y. Kim, J. H. Park, J. Kim, S. Her, Y. K. Oh, I. C. Kwon, K. Kim and S. Y. Jeong, J. Controlled Release, 2009, 135, 259–267 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra27836h |
| ‡ J. Helmlinger and C. Sengstock share first authorship. |
| This journal is © The Royal Society of Chemistry 2016 |