
Titanium dioxide nanotube arrays coated with laminin enhance C2C12 skeletal myoblast adhesion and differentiation†
Giada G. Genchi*abc,
Harald Nuhnb,
Ioannis Liakosd,
Attilio Marinoac,
Sergio Marrase,
Athanassia Athanassioud,
Virgilio Mattolia and
Tejal A. Desaib
aCenter for Micro-BioRobotics @SSSA, Istituto Italiano di Tecnologia, Viale Rinaldo Piaggio 34, 56025 Pontedera, Pisa, Italy. E-mail: giada.genchi@iit.it; Fax: +39 050883497; Tel: +39 050883035
bDepartment of Bioengineering and Therapeutic Sciences, University of California, San Francisco, 1700 4th Street, 94158, San Francisco, California, USA
cThe BioRobotics Institute, Scuola Superiore Sant'Anna, Viale Rinaldo Piaggio 34, 56025 Pontedera, Pisa, Italy
dSmart Materials, Nanophysics Department, Istituto Italiano di Tecnologia, Via Morego 30, 16163, Genoa, Italy
eNanochemistry Department, Istituto Italiano di Tecnologia, Via Morego 30, 16163, Genoa, Italy
First published on 29th January 2016
Abstract
Titanium dioxide (TiO2) nanotubes possess extraordinary yet easily tunable physico-chemical properties that have motivated a huge number of studies in various fields of nanotechnology, with recent applications even in biomedical research. Here, arrays of TiO2 nanotubes with different diameters (10 nm, 50 nm, and 100 nm) are studied in the interaction with skeletal muscle cells, in view of their use as electrically active biointerfaces. Laminin binding to nanotubes is also proposed with the purpose of enhancing nanotube biocompatibility. Laminin is indeed a protein typical of the extracellular matrix, and plays crucial roles in skeletal muscle development and activities both in vitro and in vivo. Synergistic effects of surface nanostructuring and coating on C2C12 cell responses are shown, in terms of sustained myoblast adhesion, spreading and differentiation on laminin coated nanotubes with smaller diameter, thus opening new opportunities for further exploitation of TiO2 nanotube arrays in biological environments.
1. Introduction
Since their first appearance in 1996, titanium dioxide (TiO2) nanotube arrays have undergone extensive studies for their easiness of preparation and the tunability of their geometrical and functional features. Consequently, they have found several applications in various fields of nanotechnology, for instance: photocatalysis, photovoltaics, sensing and hydrogen storage. Titanium dioxide nanotubes can be synthesized by several routes: among these, anodization of titanium foils (under either aqueous or organic electrolyte solution) at different voltages probably represents the most straightforward fabrication process, leading to the generation of highly ordered nanotube arrays with different geometrical features.1 In TiO2 nanotubes, functional properties strictly depend on both geometry (tube length, top morphology etc.)2 and post-fabrication manipulation processes. Simple thermal treatments (such as annealing in air) enable the conversion of amorphous into crystalline TiO2 nanotubes (either anatase or rutile),3 and the achievement of semimetallic conductive behavior in nanotube arrays.4 Moreover, TiO2 nanotube conductivity can be finely tuned through control of time and temperature of annealing,5 or even by doping with C, N,6 or with other transition metals.In the biomedical research, arrays of TiO2 nanotubes have been proposed for both drug delivery and tissue engineering purposes: for instance, highly porous arrays were tested for delivery of drugs such as sirolimus (immunosuppressive), paclitaxel (antitumoral)7 and enrofloxacin (antibiotic).8 For the recognized importance of surface nanostructuring of biomimetic materials in addressing cell activities and functions,9,10 TiO2 nanotubes have increasingly been proposed for interaction with several kinds of tissues.
Many investigations were done on arrays of TiO2 nanotubes with different diameters, demonstrating strikingly different effects on several cell lineages. For example, a study on adipose-derived stem cells showed significantly increased osteogenic differentiation on arrays of TiO2 nanotubes with 70 nm diameter.11 Another study on TiO2 nanotubes with 78 nm diameter demonstrated a significantly decreased inflammatory activity of macrophages after lipopolysaccharide stimulation in comparison to flat Ti surfaces.12 Nanotubes with 70–90 nm diameters were found to increase dermal fibroblast and decrease epidermal keratinocyte adhesion, proliferation and differentiation.13 Studies on TiO2 nanotube array interaction with mesenchymal stem cells (MSCs) demonstrated higher adhesion, proliferation and osteodifferentiation on 15 nm diameter nanotubes than on flat Ti surfaces and on nanotubes of higher diameter.14 Further, TiO2 nanotubes with 15–30 nm diameters were shown to promote better adhesion, proliferation, migration, and differentiation of endothelial cells,15 as well as osteoclast differentiation from hematopoietic stem cells (in comparison to nanotubes of higher diameter).16 Interestingly, bone morphogenetic protein-2 (BMP-2) coated TiO2 nanotube arrays proved to sustain higher MSC differentiation into chondrocytes when nanotube diameters were of 100 nm.17
Based on these evidences, efforts have been recently dedicated to modifying surface chemistry of TiO2 nanotube arrays with the purpose of investigating their effects on cell behavior. For instance, plasma modification with allylamine, followed by poly(sodium styrenesulfonate) adsorption or poly(ethylene glycol) binding was proposed for biomedical applications of TiO2 nanotube arrays.18 Adsorption of polydopamine coating on TiO2 nanotube arrays was also proposed and found to enhance endothelial cell attachment, proliferation, migration and release of nitric oxide, while decreasing smooth muscle cell adhesion and proliferation.19 Covalent binding with (3-aminopropyl)trimethoxysilane was proposed for interaction with fibroblasts, supporting cell adhesion and proliferation.20 Immobilization of Arg-Gly-Asp (RGD) cell adhesive peptides was demonstrated on TiO2 nanotubes upon silanization and following reaction with the heterobifunctional cross-linker 3-succinimidyl-3-maleimido propionate.21 Finally, further chemical modifications for TiO2 nanotubes relying on (3-aminopropyl)triethoxysilane, carbonyldiimidazole and 11-hydroxyundecylphosphonic acid were proposed in an attempt at developing biosensors.22 Nonetheless, just a few studies explored synergistic effects of surface nanotopography and chemistry on cell behavior.17,23–25 Wide opportunities thus remain for the application of TiO2 nanotube arrays (both uncoated and coated) in different contexts of tissue engineering.
Despite the huge interest on titanium dioxide nanostructures, TiO2 nanotube arrays have never been tested with skeletal muscle, which would highly benefit from the interaction with a topographically and electrically active interface, when necessary. As a proof of concept, arrays of TiO2 nanotubes with different diameters were thus prepared and modified to test their suitability to skeletal myoblast culture. Arrays were obtained by anodization of titanium foils at different voltages against Pt under organic electrolyte solution (with ammonium fluoride and ethylene glycol), and annealed to obtain crystalline TiO2 nanotubes. Moreover, silanization with (3-aminopropyl)triethoxysilane and laminin binding mediated by genipin were sequentially performed to modify surface chemistry. Upon achievement of different nanotube diameters and surface chemistries, simultaneous investigations on array nanotopography (same morphology, but different size of geometrical nanofeatures) and on possible additive effects of laminin coating over TiO2 nanotube array bioactivity could be performed. In the following, the relevance of nanotube diameter to C2C12 biological activities is shown. The effectiveness of surface coating in synergy with nanotopography to improve myoblast adhesion and differentiation is also shown. The goal of this study is to support the use of titanium dioxide nanotube arrays as cell/tissue instructive interfaces for both skeletal muscle engineering in vitro (in the context of tissue transplantation procedures), and for skeletal muscle stimulation in vivo (through implantable devices).
2. Methods
2.1. Substrate preparation
TiO2 nanotube arrays were prepared by anodization of titanium foils (2 cm2, Sigma) against Pt electrodes (2 cm2), under a 0.3% ammonium fluoride (Sigma) solution in ethylene glycol (Sigma): pure water (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v, Milli-Q Millipore). Different voltages (4 V, 20 V, and 40 V) were applied to the titanium foils and platinum electrodes, held parallel and at a distance of 1 cm while immersed for 18 mm in the anodization solution. Reaction temperature was kept constant to 25 °C in a water jacketed reaction chamber, and nanotube growth was allowed for 4 h at 4 V, and for 2 h at 20 V and 40 V. Nanotubes with different diameters were thus obtained on titanium foils and were used to establish their effect on myoblast behavior. After anodization, samples were cleaned by sonication for 20 min under ethanol, then they were annealed in air at 400 °C for 1 h to remove fluoride contaminants and convert amorphous titanium dioxide into anatase.3,26
:1 v/v, Milli-Q Millipore). Different voltages (4 V, 20 V, and 40 V) were applied to the titanium foils and platinum electrodes, held parallel and at a distance of 1 cm while immersed for 18 mm in the anodization solution. Reaction temperature was kept constant to 25 °C in a water jacketed reaction chamber, and nanotube growth was allowed for 4 h at 4 V, and for 2 h at 20 V and 40 V. Nanotubes with different diameters were thus obtained on titanium foils and were used to establish their effect on myoblast behavior. After anodization, samples were cleaned by sonication for 20 min under ethanol, then they were annealed in air at 400 °C for 1 h to remove fluoride contaminants and convert amorphous titanium dioxide into anatase.3,26
Soon after annealing, samples were incubated with a 60% ethanol, 20% (3-aminopropyl)triethoxysilane (APTES, Sigma), 1.2% NH3 water solution (pH = 9) for 4 h at 80 °C, and then with a 5 mg ml−1 genipin (Sigma) solution in pure water for 60 min at 37 °C. Surface chemistry was finally modified by incubation with a 50 μg ml−1 laminin (Life Technologies) solution (pH = 4) at 37 °C for 12 h in 20 mM sodium acetate and 1 mM calcium chloride to promote both laminin precipitation under biomimetic conditions and laminin binding to genipin.26–28 Genipin was used to mediate crosslinking of the protein as detailed elsewhere.27 Surface composition of native titanium foils is schematized in Fig. 3a, whereas silanization of the surface and genipin/laminin binding are depicted in Fig. 3b–d. Binding of laminin to the surface was considered completed upon development of blue color on the samples (see Fig. 3e, right end).
Substrates were cut into 25 mm2 pieces for biological study. As purchased titanium foils cut into 25 mm2 pieces after annealing were considered as control substrates. All of the samples were sterilized prior to cell culture by incubation with a 1× penicillin/streptomycin (Life Technologies) solution in phosphate-buffered saline (1× PBS, Life Technologies).
2.2. Substrate characterization
Structural characterization of samples was carried out with X-ray diffraction (XRD) analysis. XRD patterns were recorded on a PANalytical Empyrean X-ray diffractometer equipped with a 1.8 kW CuKα ceramic X-ray tube, PIXcel3D 2 × 2 area detector and operating at 45 kV and 40 mA. The diffraction patterns were collected in air at room temperature by using Parallel-Beam (PB) geometry and symmetric reflection mode. XRD data analysis was carried out by using HighScore 4.1 software from PANalytical.Surface morphology of substrates was characterized with a scanning electron microscope (Helios 600i, FEI) by applying 10 kV acceleration and 86 pA current. Prior to imaging, samples were made conductive by Au sputtering (25 mA current, Q150 R Quorum sputterer) for 60 s.
Surface chemistry of substrates was analyzed with X-ray photoelectron spectroscopy (XPS). Spectra were acquired with a spectrometer (Specs Lab2) equipped with a monochromatic Mg Kα X-ray source (1253 eV radiation) and a Phoibos analyzer Has 3500 (Emispherical Energy Analyzer). The following parameters were used: 7.5 kV voltage and 9.5 mA current (∼2 × 10−9 mbar pressure). Small area lens mode was used for both wide and narrow scans. For the wide scans, the energy pass was set at 90 eV, the energy step at 0.5 eV, and the scan number at 1. For the narrow high-resolution scans, the pass energy was set at 30 eV, the energy step at 0.2 eV and the scan number at 10. Collected spectra were analyzed using CasaXPS software.
2.3. Cell cultures
For proliferation studies, C2C12 myoblasts (CRL 1772, ATCC) were seeded at a density of 10000 cells per cm2 over both uncoated and laminin coated substrates. Cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Life Technologies), supplemented with 10% fetal bovine serum (FBS, Life Technologies) and 0.1 mg ml−1 gentamicin (Life Technologies). Medium was changed every two days.
For differentiation studies, C2C12 myoblast were seeded at a density of 30000 cells per cm2 over both uncoated and laminin coated substrates. Adhesion was allowed for 24 h from seeding, then myoblast differentiation into myotubes was induced by supplying confluent cells with DMEM supplemented with 1% FBS, 1% ITS (bovine insulin, human transferrin, sodium selenite and ethanolamine, Sigma), and 0.1 mg ml−1 gentamicin. Medium was daily changed for three days after differentiation induction. In both studies, cells were maintained at 37 °C in a 5% CO2 saturated humidity atmosphere.
2.4. Biological testing
Myoblast adhesion on both uncoated and laminin coated nanotube arrays was investigated with fluorescence staining of vinculin, actin and nuclei after 24 h from cell seeding. Samples were first washed with PBS, and then fixed with 4% paraformaldehyde in PBS at 4 °C for 20 min. To suppress aspecific fluorescence, samples were incubated with 1 mg ml−1 sodium borohydryde (Sigma) in PBS for 20 min, then permeabilization of cell membranes was performed with 0.1% Triton X100 (Dow Chemical) in PBS for 15 min. Saturation of aspecific binding sites for antibodies was performed with 10% goat serum (GS, Life Technologies) at 37 °C for 1 h. Incubation with a rabbit primary antibody (1:100 diluted IgG in 10% GS, Millipore) against vinculin was performed for 45 min at 37 °C, then samples were incubated with a 10% GS solution containing a mouse antirabbit secondary antibody (1:250 diluted IgG, Millipore), tetramethylrhodamine phalloidin (TRITC-phalloidin, 100 μM diluted, Millipore) and 4′,6-diamidino-2-phenylindole (DAPI, 1 μM diluted, Millipore) at room temperature for 30 min. Unbound and weakly bound antibodies were removed by washes with 0.45 M NaCl PBS and PBS. Images of the cultures were taken with an inverted confocal microscope (C2s, Nikon). The number of vinculin clusters per cell on each substrate was obtained with automated counting through ImageJ and its “Find Maxima” process (more details on the procedure are presented in the ESI†). For statistical purposes, ten images were considered per substrate.
Myoblast viability and capability to cover both the uncoated and the laminin coated substrates were investigated after three days of culture under proliferative conditions. Live/Dead® staining (Molecular Probes) was performed following the manufacturer's instructions. Briefly, myoblasts were incubated with cell culture medium added with 2 μM calcein AM and 4 μM EthD-1 at 37 °C in the dark for 10 min, then cells were washed with PBS and immediately imaged with an inverted epifluorescence microscope (Eclipse TI, Nikon). Cell coverage was quantified with free ImageJ software (http://rsbweb.nih.gov/ij/) on five low magnification images of the cultures.
Myoblast metabolic activity on both uncoated and coated samples was quantified with WST-1 assay (BioVision) after 24 and 72 h of culture under proliferative conditions. Cell cultures were treated with 300 μl of culture medium added with 30 μl of WST-1 solution for 2 h, then the absorbance of the supernatants was read at 450 nm with a microplate reader (Victor 3, Perkin Elmer).
Myoblast proliferative capability on both uncoated and coated samples was quantified with PicoGreen assay (Thermo Scientific) after 24 and 72 h of culture on the same samples that underwent WST-1 assay. Samples were first washed with PBS, then were lysed with 500 μl of ultrapure water and three cycles of freezing/thawing. The assay (that measures the quantity of ds-DNA in solution) was performed following the manufacturer's instructions. Briefly, 100 μl of working solution were mixed to 50 μl of lysate, and then 150 μl of solution containing the PicoGreen dye were added. Samples were incubated in the dark for 10 min, and finally fluorescence was read at an emission wavelength of 535 nm (by using a 485 nm excitation wavelength with the microplate reader).
Myoblast capability to fuse into myotubes was investigated on fixed cultures upon fluorescence staining of actin and nuclei with TRITC-phalloidin and DAPI, respectively, three days after differentiation induction with the low-serum, ITS-added culture medium. Images of the cultures were taken with epifluorescence microscope, and myotube width was measured with the “Line” tool of ImageJ. For statistical purposes, over 300 myotubes were considered per substrate.
2.5. Statistical analysis
Data from quantification of number of vinculin clusters per cell, of substrate coverage by myoblasts, of myoblast metabolic activity and proliferation are presented as mean ± standard deviation. Data from quantification of myotube widths are presented as median ± 95% confidence interval in box-plots. Upon verification of data normality with Shapiro test, ANOVA was performed, followed by Tukey's HSD post-hoc test. All experiments were carried out in triplicate.3. Results and discussion
Scanning electron microscopy (SEM) images of the 50 nm diameter nanotube array (as a representative sample) are reported in Fig. 1. After sample anodization and cleaning, highly ordered, open-top nanotubes were achieved (Fig. 1a, left). After annealing, array morphology was fully retained (Fig. 1a, right).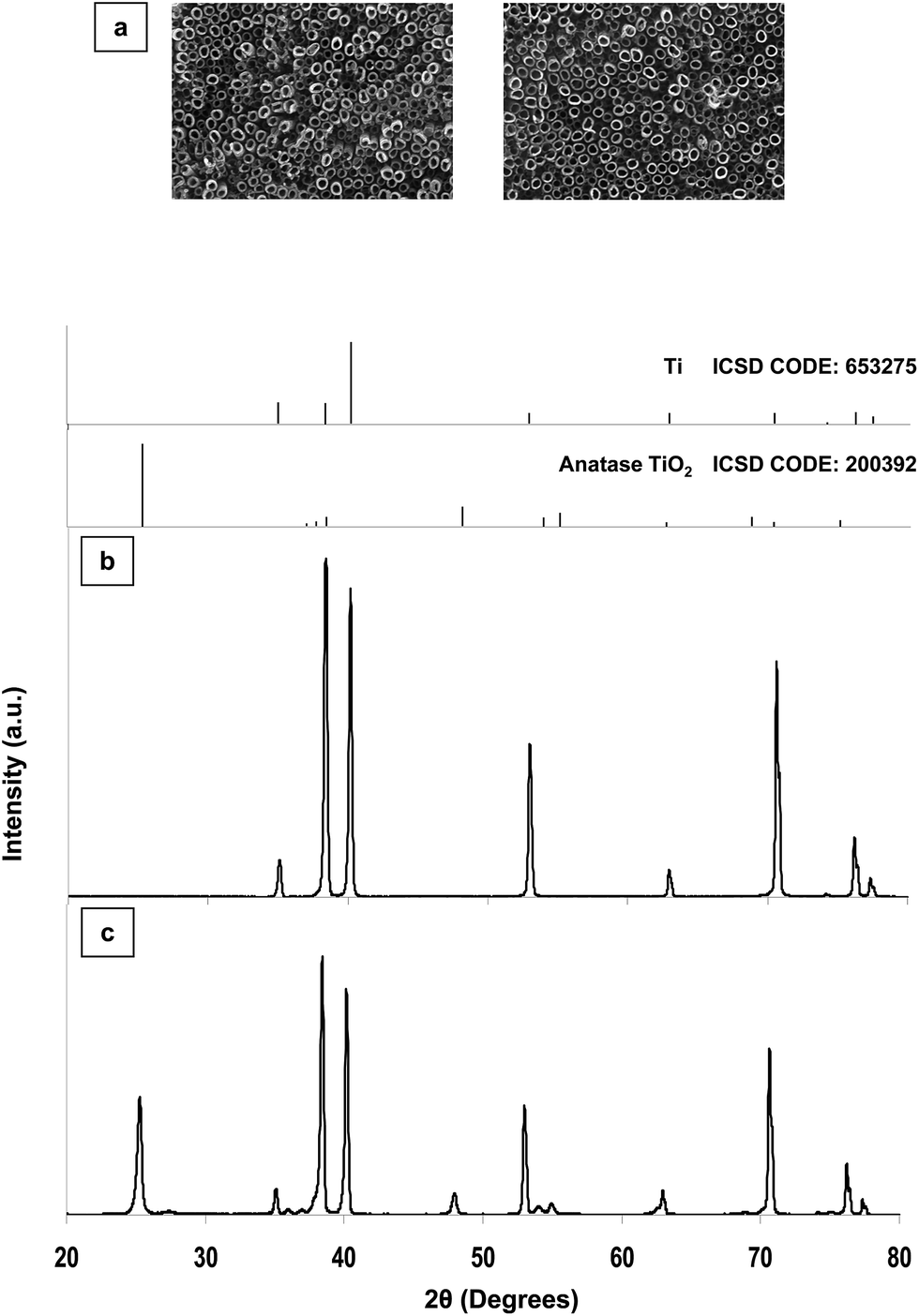 | ||
| Fig. 1 Scanning electron microscopy images (a) of a representative array of TiO2 nanotubes (50 nm diameter) soon after anodization and cleaning (left) and after annealing (right). X-ray diffraction patterns of the same array soon after anodization and cleaning (b) and after annealing (c). | ||
X-ray diffraction (XRD) patterns of the 50 nm diameter nanotube array (as a representative sample) are also reported in Fig. 1. The XRD pattern of the sample soon after anodization and cleaning (Fig. 1b) shows no peaks besides those of metallic Ti, whereas the XRD pattern of the same sample after annealing (Fig. 1c) shows the appearance of other two peaks: one intense peak at 25.3° and another one at 48.2°, demonstrating the conversion of amorphous TiO2 into anatase with annealing at 400 °C in air.
X-ray photoelectron spectroscopy (XPS) analysis of the 50 nm diameter nanotube array (as a representative sample) is reported in Fig. 2. The wide spectrum of the sample after anodization and annealing (Fig. 2a) shows peaks of elements expected in the array composition, that is the Ti2p peak (at 458.0 eV) and the O1s peak (at 530.0 eV), but also peaks of residual contaminants from the anodization solution: the C1s peak (at 285.0 eV) and the F1s peak (at 685.0 eV). The latter peak disappears after annealing (Fig. 2b), whereas the C1s peak persists maybe due to organic volatile contaminants. Fig. 2c reports the deconvolution of the O1s peak of the sample soon after anodization and cleaning, whereas Fig. 2d that one of the sample after annealing. The decrease of the O1s peak component at higher binding energy in Fig. 2d compared to 2c provides evidence of the conversion of the amorphous phase into the anatase phase after annealing,26 in agreement with the XRD data. Fig. S1 (in ESI†) demonstrates that the C1s peak occurs at the same binding energy before (Fig. S1a†) and after (Fig. S1b†) annealing, thus confirming that the effect on the O1s peak component at higher binding energy was only due to annealing and not to charging of the sample during XPS analysis.
 | ||
| Fig. 2 X-ray photoelectron spectroscopy analysis of a representative array of TiO2 nanotubes (50 nm diameter) soon after anodization and cleaning (a and c) and after annealing (b and d). Arrows in the narrow spectra (c and d) evidence the decrease of the O1s peak component at higher binding energy. | ||
Laminin coating procedures involving genipin are schematized in Fig. 3a–d. They macroscopically resulted for all samples in a neat variation of the substrate color from grey to blue (Fig. 3e, array of nanotubes with 100 nm diameter as a representative example).
 | ||
| Fig. 3 Schematization of the laminin binding procedures performed with genipin (as cross-linking agent) and laminin (as cell pro-adhesive and pro-differentiative protein) on bare titanium foil and TiO2 nanotube arrays (a–d). Macroscopic images of a representative substrate (array of nanotubes with 100 nm diameter) before and after laminin coating (e). | ||
After sample anodization at different voltages and annealing, the achievement of arrays with different nanotube diameters was confirmed with SEM (Fig. 4). Nanotube diameters were significantly different, being the average inner diameter of 10 ± 1 nm on 4 V samples, of 50 ± 10 nm on 20 V samples, and of 100 ± 10 nm on 40 V samples. The average outer diameter was 20 ± 1 nm on 4 V samples, 80 ± 10 nm on 20 V samples, and 140 ± 10 nm on 40 V samples.
 | ||
| Fig. 4 Scanning electron microscopy images of anodized and annealed samples before and after laminin coating. All scale bars are 300 nm. | ||
After laminin binding, surface nanostructuring was well retained on the coated arrays, in particular with the nanotubes of higher diameters (Fig. 4). The coating appeared to be more homogenous as the nanotube diameter decreased. On arrays of nanotubes with 10 nm diameter, nanotube tops were completely closed by the protein layer. SEM imaging of the control sample before and after annealing can be found in Fig. S2 of ESI.†
XPS was also performed on the 50 nm diameter nanotube array (as a representative sample) at the end of the laminin binding procedures. The wide spectrum (Fig. 5a) shows the significant decrease of the Ti2p peak due to the protein coating, and the appearance of the Si2p peak (at 102.0 eV) and N1s peak (at 400.0 eV) as a consequence of the incubations with silane, genipin and protein.
 | ||
| Fig. 5 X-ray photoelectron spectroscopy analysis of a representative array of TiO2 nanotubes (50 nm diameter). Wide spectrum of the array at the end of the laminin binding procedure (a). Deconvolution of the C1s peak after annealing (b), after silanization (c), after incubation with genipin (d), and with laminin (e). | ||
Fig. 5b–e show the deconvolution of the C1s peak of the array at the beginning and after each step of the laminin binding procedures. Fig. 5b shows the presence of a C1s peak in the native array, very likely due to a surface contamination from volatile organic compounds. Fig. 3c shows that, after silanization, the C1s peak presents two components: one peak at 285.0 eV, also ascribable to the C–H and C–C bonds in the hydrocarbon chain of APTES, and another peak at higher binding energy (286.6 eV) mainly arising from the C–N bond of APTES. Fig. 3d shows that, after incubation with genipin, the C1s peak also presents a large component at 286.7 eV ascribable to the C–OH and C–N bonds of genipin, and two components due to the α carbon (at 288.8 eV) and β carbon (at 285.4 eV) of the ester group of genipin. Fig. 3e shows that, after incubation with laminin, the C1s peak also presents a very high peak at 288.2 eV arising from peptide bonds.
Cell adhesion was investigated after 24 h of proliferation on the substrates. To this purpose, fluorescent staining of vinculin, cytoskeletal actin and nuclei was performed. Fig. 6 shows vinculin stained in green, actin stained in red and nuclei stained in blue. In Fig. 6a, all samples show vinculin clusters, mainly localized at the cell edges. Fluorescent staining of vinculin is also present with perinuclear localization in the cytoplasm of cells on arrays of nanotubes with 10 nm and 100 nm diameter, as well as on coated controls. Fig. 6b shows the quantification of vinculin clusters per cell. The complete statistical analysis of these data is reported in Table S1,† where * means p < 0.05, ** mean p < 0.005, and *** mean p < 0.001. The number of vinculin clusters per cell was 8.8 ± 1.2 on the uncoated control substrates, 8.8 ± 1.2, 7.3 ± 1.2, and 7.8 ± 0.7 on uncoated arrays of nanotubes with 10 nm, 50 nm and 100 nm diameter, respectively. This number was comparable on all of the uncoated substrates. The number of vinculin clusters per cell was 12.3 ± 1.2 on the coated control substrates, 15.1 ± 1.6, 12.5 ± 0.5, and 10.0 ± 1.7 on coated arrays of nanotubes with 10 nm, 50 nm and 100 nm diameter, respectively. The laminin coated arrays with 10 nm diameter nanotubes supported a significantly higher number of vinculin clusters compared to the coated control substrates. In all cases, laminin coated substrates supported a significantly higher number of vinculin clusters than the uncoated arrays with the same nanotube diameter.
 | ||
| Fig. 6 Confocal fluorescence microscopy images of C2C12 myoblasts after 24 h of culture under proliferative conditions on both uncoated and laminin coated samples (a). Vinculin is stained in green; cytoskeletal actin in red; nuclei in blue. Quantification of the number of vinculin clusters per cell on both uncoated and laminin coated samples (b). The complete statistical analysis is reported in ESI Table S1.† | ||
Viability was also investigated with Live/Dead® staining. Fig. 7a shows fluorescence microscopy images of the cultures after 72 h from seeding on both uncoated and laminin coated samples. Live cells are stained in green, whereas dead cells are stained in red. Fig. 7a clearly demonstrates a well sustained cell viability on all of the samples, where dead cells are present in a very negligible number (<1%). Furthermore, myoblasts appear as homogeneously covering all of the substrates. Fluorescence microscopy images were used to quantify the extent of surface coverage with ImageJ software, and results are reported in Fig. 7b. The complete statistical analysis of these data is reported in Table S2.† Percentage area covered by cells was 47 ± 2% on the uncoated control substrates, 49 ± 1%, 36 ± 1%, and 41 ± 1% on uncoated arrays of nanotubes with 10 nm, 50 nm and 100 nm diameter, respectively. Therefore, it was comparable on all of the uncoated substrates except for arrays of nanotubes with 50 nm diameter, where it was significantly lower. Percentage area covered by cells was 62 ± 2% on the coated control substrates, 76 ± 1%, 56 ± 2%, and 68 ± 4% on coated arrays of nanotubes with 10 nm, 50 and 100 nm diameter, respectively. Therefore, it was significantly higher on coated arrays of nanotubes with 10 nm diameter in comparison to the coated control substrates and to coated arrays of nanotubes with 50 nm diameter.
 | ||
| Fig. 7 Fluorescence microscopy images of C2C12 myoblasts incubated with Live/Dead® staining after three days of culture under proliferative conditions on uncoated and laminin coated TiO2 nanotube arrays (a). C2C12 myoblast coverage of uncoated and laminin coated TiO2 nanotube arrays (b). Percentage area measured with ImageJ software on fluorescence microscopy images after Live/Dead® staining. The complete statistical analysis is reported in ESI Table S2.† | ||
Cell metabolism was investigated with WST-1 assay after 24 and 72 h since seeding on the substrates. Results are reported in Fig. 8a and b, and their complete statistical analysis is presented in Table S3.† After 24 h of culture, absorbance was 0.069 ± 0.018, 0.107 ± 0.005, 0.102 ± 0.004, and 0.110 ± 0.011 for uncoated control substrates, 10 nm, 50 nm and 100 nm diameter nanotube arrays, respectively. Absorbance was 0.085 ± 0.002, 0.106 ± 0.005, 0.103 ± 0.004, 0.107 ± 0.011 for coated control substrates, 10 nm, 50 nm and 100 nm diameter nanotube arrays, respectively. After 24 h of culture on uncoated substrates, a significantly higher metabolic activity was shown by cells on 50 nm and 100 nm diameter nanotubes compared to controls. After 24 h of culture on laminin coated substrates, a significantly higher metabolism was shown by cells on 10 nm and 100 nm diameter nanotubes compared to controls. After 72 h of culture, absorbance was 0.608 ± 0.059, 0.431 ± 0.058, 0.566 ± 0.017, 0.517 ± 0.049 for uncoated control substrates, 10 nm, 50 nm and 100 nm diameter nanotube arrays, respectively. Absorbance was 0.249 ± 0.082, 0.204 ± 0.024, 0.333 ± 0.014, 0.310 ± 0.045 for coated control substrates, 10 nm, 50 nm and 100 nm diameter nanotube arrays, respectively. After 72 h of culture on uncoated substrates, a significantly lower metabolism was shown by cells on 10 nm and 100 nm diameter nanotubes compared to controls. After 72 h of culture on laminin coated substrates, no significant differences were found in metabolism of cells cultured on different substrates. In all cases, laminin coated substrates supported a significantly lower cell metabolism than the uncoated arrays with the same nanotube diameter.
 | ||
| Fig. 8 Quantification of C2C12 metabolic activity during proliferation through WST-1 assay after 24 h (a) and 72 h (b) from seeding. Quantification of cell proliferation through PicoGreen assay after 24 h (c) and 72 h (d) from seeding. The complete statistical analysis is reported in ESI Tables S3 and S4.† | ||
Cell proliferation was quantified with PicoGreen assay after 24 and 72 h since seeding on the substrates. Results are reported in Fig. 8c and d, and their complete statistical analysis is presented in Table S4.† After 24 h of culture, fluorescence from ds-DNA was (72 ± 3, 89 ± 5, 82 ± 8, 74 ± 4) × 103 for uncoated control substrates, 10 nm, 50 nm and 100 nm diameter nanotube arrays, respectively. Fluorescence was (76 ± 2, 98 ± 8, 74 ± 5, 115 ± 4) × 103 for coated control substrates, 10 nm, 50 nm and 100 nm diameter nanotube arrays, respectively. After 24 h of culture on uncoated substrates, a significantly higher fluorescence was obtained from cells on 10 nm diameter nanotubes compared to controls. After 24 h of culture on laminin coated substrates, a significantly higher fluorescence was obtained from cells on 10 nm and 100 nm diameter nanotubes compared to controls. Fluorescence was significantly higher on laminin coated arrays with 100 nm diameter nanotubes in comparison to the uncoated arrays with 100 nm diameter nanotubes. After 72 h of culture, fluorescence was (70 ± 6, 58 ± 11, 74 ± 8, 43 ± 12) × 104 for uncoated control substrates, 10 nm, 50 nm and 100 nm diameter nanotube arrays, respectively. Fluorescence was (31 ± 1, 21 ± 1, 41 ± 6, 67 ± 1) × 104 for coated control substrates, 10 nm, 50 nm and 100 nm diameter nanotube arrays, respectively. After 72 h of culture on uncoated substrates, a significantly lower fluorescence was obtained from cells on 100 nm diameter nanotube arrays compared to controls. After 72 h of culture on laminin coated substrates, a significantly higher fluorescence was obtained from cells on 100 nm diameter nanotube arrays compared to controls. Fluorescence was significantly lower on coated controls, 10 nm and 50 nm diameter nanotube arrays compared to the uncoated counterparts, whereas it was significantly higher on 100 nm diameter nanotube arrays. Fluorescence from ds-DNA content data therefore demonstrate a high coherence to cell metabolism data.
Differentiation was also investigated with fluorescent staining of cytoskeletal actin and nuclei. Fig. 8 shows fluorescence microscopy images of cultures after three days of differentiation on TiO2 nanotube arrays. All of the substrates support C2C12 myoblast differentiation in elongated and multinucleated myotubes, although uncoated controls retained a consistent number of single round, undifferentiated cells. ImageJ analysis was performed on fluorescence microscopy images to extract myotube widths. Median myotube widths were calculated and reported with their confidence intervals at 95% in Fig. 9. Median myotube widths are: 8 μm on uncoated control substrates, 11 μm, 10 μm, and 11 μm on uncoated arrays of nanotubes with 10 nm, 50 nm and 100 nm diameter, respectively. The complete statistical analysis of these data is reported in Table S5.† Myotube widths on arrays of nanotubes with 10 nm and 100 nm diameter are significantly higher than on the control substrates and on arrays of nanotubes with 50 nm diameter. Median myotube widths are also: 9 μm on coated control substrates, 16 μm, 11 μm, and 11 μm on coated arrays of nanotubes with 10 nm, 50 nm and 100 nm diameter, respectively. Thus, the increase of myotube width on coated arrays of nanotubes with 10 nm diameter compared to all of the other coated substrates is highly significant (Fig. 10).
 | ||
| Fig. 9 Fluorescence microscopy images of C2C12 myoblasts differentiated into myotubes on both uncoated and laminin coated TiO2 nanotube arrays after three days from differentiation induction. Cytoskeletal actin is stained in red; nuclei, in blue. | ||
 | ||
| Fig. 10 Box plot of myotube widths (median and 95% confidence intervals) for differentiated C2C12 cultures on both uncoated and laminin coated TiO2 nanotube arrays. For complete statistical analysis, the Reader is referred to ESI Table S5.† | ||
A great body of evidences in the literature supports the use of titanium oxide materials for several technological applications, among which biomedical device development. In the context of skeletal muscle tissue engineering, an oxygen-rich coating on titanium oxide substrates was investigated demonstrating enhanced myoblast adhesion and proliferation.29 Also, titanium dioxide nanorods were prepared by sol–gel electrospinning and used with C2C12 myoblasts, supporting adequate cell adhesion, spreading and proliferation.30 Often prepared by anodization under electrolytic solutions, arrays of titanium dioxide nanotubes were tested with several cell lineages showing enhanced cell adhesion and differentiation,11,13–17,19,20,23–25 but to date evidences of their potentialities to support skeletal muscle cell growth are still missing.
Here we investigated the effects of arrays of TiO2 nanotubes on skeletal muscle cell behavior in view of a possible use of arrays as electrically active interfaces for muscle growth and stimulation. Arrays of titanium dioxide nanotubes can indeed easily be prepared with finely tuned features (i.e. different nanotube diameters, lengths, wall thicknesses etc.), and their bulk properties can be simply varied for example by thermal treatment to show improved electric properties.31–33 Moreover, their surface chemistry can efficiently be modified to improve functional properties25,34 and biocompatibility.35,36 Nonetheless, few studies in the literature show synergistic effects of nanotube diameter and coating on cell differentiation: MSCs for example undergo enhanced osteogenic differentiation on arrays of TiO2 nanotubes with 15 nm diameter after coating with BMP-2.17 Other authors reported 30% faster osteogenic differentiation of human osteoblasts on tantalum coated arrays of TiO2 nanotubes with 100 nm diameter in comparison to the uncoated ones.26 In this paper, we proposed modification of TiO2 nanotube arrays with laminin to evaluate possible synergistic effects of surface nanotopography and different surface chemistry on C2C12 myoblast proliferative and differentiative behavior. Abundant in the basement membrane of skeletal muscle where accomplishes important functional role,37 laminin is indeed a protein well known to exert pro-adhesive and pro-differentiative effect on skeletal myoblasts also in vitro.38,39 Here, laminin was covalently bound to the surface of titanium nanotube arrays to prevent common washout of simply adsorbed biomolecules from TiO2 array surface.17 To this purpose, samples were silanized under basic solution at high temperature, incubated with genipin (acting as a bifunctional cross-linking agent), and then incubated with an acidic solution containing the protein. Silanization at high temperature was done for properly oriented binding of APTES to pendant hydroxyl groups on titanium dioxide nanotubes, leaving free amine groups for further reaction.26 Acidic incubation was then necessary to genipin ring opening, and directional binding to protein amine residues.40 In a previous study, these procedures were demonstrated to be effective at binding collagen on silicone substrates for long term culture of H9c2 cells.27 In the present study, these data were merged to the information that an acidic solution of laminin determines protein arrangement and precipitation with a structure highly similar to the one in vivo,28 with the purpose of conferring enhanced bioactivity to TiO2 nanotube arrays.
At the end of the laminin binding procedure, a conformal layer of protein was found on all nanotube arrays, and homogeneity of the laminin coating was found to be increasing as the nanotube diameter decreased. High homogeneity of the laminin coating was also found on control substrates. In the literature, full retention of surface morphology was obviously reported only for inorganic coatings24,34 and for coating with hexapeptides on 100 nm diameter nanotube arrays.23 In this work, difference in protein coating homogeneity did not affect focal adhesion development. Indeed, vinculin clusters could be found on all of the substrates after 24 h of proliferation (also on the uncoated ones). Nonetheless, the number of vinculin clusters per cell was comparable on the uncoated substrates, whereas it was significantly higher on the 10 nm diameter nanotubes compared to all of the other coated substrates, including controls. This also demonstrates that the laminin coating significantly increased the number of vinculin clusters per cell, and thus cell adhesion, but only in synergy to substrate topography. These results are in line with another work in the literature showing that vinculin expression (evaluated in pixel number on confocal images) per cell was significantly increased by an oxygen-rich coating from molten TiO2 nanoparticles on microroughened titanium surfaces in comparison to uncoated surfaces.29
Similarly to immunostaining data, quantification of cell coverage demonstrated that the laminin coated arrays of nanotubes with 10 nm diameter better supported C2C12 adhesion than the other substrates. Comparable results were also achieved on laminin coated arrays of nanotubes with 100 nm diameter. All of the samples supported comparable high viability. Proliferation data achieved from cell metabolism and ds-DNA quantification show that the laminin coated array of nanotubes with 10 nm diameter are the substrates mostly influencing cell behavior. Collectively, our results of myoblast behavior under proliferative conditions on titanium dioxide nanotube arrays are slightly different from what was found by some authors with other cells, that is (1) neat dependence on nanotube diameter,14,15 and (2) prevalent responsiveness to surface topography than to surface chemistry.35 In the case of both human and rat MSCs, better adhesion was for instance shown on 15 nm diameter nanotube arrays than on 100 nm nanotube arrays despite coating with BMP-2.17 Almost 70% of rat MSCs underwent apoptosis after 48 h of culture over 100 nm diameter nanotube arrays,14 whereas ∼40% of human MSC cultures were found apoptotic on 100 nm diameter nanotube arrays. When the latter were coated with BMP-2, only 10% of human MSCs underwent apoptosis.17 In the case of human nasal epithelial cell, better adhesion and viability were instead shown on 25 nm diameter compared to 100 nm diameter nanotubes, both in the presence and in the absence of electron beam deposited Ag coating.25 Instead, our results of C2C12 proliferation show a non-obvious linear dependence on nanotube diameter, both in the presence and in the absence of surface coating. Further studies are therefore necessary to elucidate why C2C12 myoblasts are negatively influenced by 50 nm diameter nanotube arrays.
Improved C2C12 differentiation was observed on arrays of nanotubes with 10 nm diameter in comparison to the other substrates, but only after laminin coating. Interestingly, past study on adult rat ventricular cardiomyocytes demonstrated their ability to retain the differentiated phenotype on titanium dioxide ceramic disks coated with a sol–gel derived titanium dioxide layer.41
All of these evidences therefore demonstrate that several investigations have to be performed with different cell types and culture conditions to get final indications on the potentialities of arrays of TiO2 nanotubes with different diameter/surface chemistry for tissue engineering purposes. Although preliminary, our results suggest that arrays of titanium dioxide nanotubes with 10 nm diameter can represent useful interfaces for interaction with skeletal muscle cells. Improvements in the design of these interfaces are obviously necessary and may consist for instance in matching their mechanical properties to the target tissue. In this concern, promising evidences on implementation of TiO2 nanotube arrays in flexible nanohybrid devices have recently started to emerge.42
4. Conclusions
Skeletal muscle cell response to arrays of titanium dioxide nanotubes with different diameters and surface chemistries was investigated. Improved cell adhesion and differentiation were found on arrays of nanotubes with the smaller diameter (10 nm) after coating with laminin, a pro-adhesive and pro-differentiative protein typical of the extracellular matrix. These results provide promising evidences of the strong interplay of both material nanotopography and surface chemistry in affecting cell behavior. Further studies are obviously necessary to better elucidate differentiation of skeletal muscle cells on arrays of titanium dioxide nanotubes, as well as functional behavior upon generation of electrical currents.Acknowledgements
Mr Valfredo Zolesi (Kayser Italia S.r.l.) is gratefully acknowledged for funding Giada Graziana Genchi's doctoral studies. Dr Clive Hayzelden (San Francisco State University) is acknowledged for technical advice in SEM imaging. Dr Gianni Ciofani (Istituto Italiano di Tecnologia) is also gratefully acknowledged for technical advice in biological assays and for support in data analysis.Notes and references
- K. Lee and P. Schmuki, Electrochim. Acta, 2013, 100, 229–235 CrossRef CAS
.
- A. Mazzarolo, K. Lee, A. Vicenzo and P. Schmuki, Electrochem. Commun., 2012, 22, 162–165 CrossRef CAS
- A. Ghicov, H. Tsuchiya, J. M. Macak and P. Schmuki, Phys. Status Solidi, 2006, 203, R28–R30 CrossRef CAS
- R. Hahn, F. Schmidt-Stein, J. Salonen, S. Thiemann, Y. Song, J. Kunze, V.-P. Lehto and P. Schmuki, Angew. Chem., Int. Ed., 2009, 48, 7236–7239 CrossRef CAS PubMed
- A. Tighineanu, T. Ruff, S. Albu, R. Hahn and P. Schmuki, Chem. Phys. Lett., 2010, 494, 260–263 CrossRef CAS
- R. P. Vitiello, J. M. Macak, A. Ghicov, H. Tsuchiya, L. F. P. Dick and P. Schmuki, Electrochem. Commun., 2006, 8, 544–548 CrossRef CAS
- L. Peng, A. D. Mendelsohn, T. J. LaTempa, S. Yoriya, C. G. Grimes and T. A. Desai, Nano Lett., 2009, 9, 1932–1936 CrossRef CAS PubMed
- P. Huang, J. Wang, S. Lai, F. Liu, N. Ni, Q. Cao, W. Liu, D. Y. B. Deng and W. Zhou, J. Mater. Chem. B, 2014, 2, 8616–8625 RSC
- K. von der Mark, J. Park, S. Bauer and P. Schmuki, Cell Tissue Res., 2010, 339, 131–153 CrossRef CAS PubMed
- S. Bauer, P. Schmuki, K. von der Mark and J. Park, Prog. Mater. Sci., 2013, 58, 261–326 CrossRef CAS
- L. Lv, Y. Liu, P. Zhang, X. Zhang, J. Liu, T. Chen, P. Su, H. Li and Y. Zhou, Biomaterials, 2015, 39, 193–205 CrossRef CAS PubMed
- P. Neacsu, A. Mazare, A. Cimpeana, J. Park, M. Costache, P. Schmuki and I. Demetrescu, Int. J. Biochem. Cell Biol., 2014, 55, 187–195 CrossRef CAS PubMed
- B. S. Smith, S. Yoriya, T. Johnson and K. C. Popat, Acta Biomater., 2011, 7, 2686–2696 CrossRef CAS PubMed
- J. Park, S. Bauer, K. von der Mark and P. Schmuki, Nano Lett., 2007, 7, 1686–1691 CrossRef CAS PubMed
- J. Park, S. Bauer, P. Schmuki and K. von der Mark, Nano Lett., 2009, 9, 3157–3164 CrossRef CAS PubMed
- J. Park, S. Bauer, K. A. Schlegel, F. W. Neukam, K. von der Mark and P. Schmuki, Small, 2009, 5, 666–671 CrossRef CAS PubMed
- J. Park, S. Bauer, A. Pittrof, M. S. Killian, P. Schmuki and K. von der Mark, Small, 2012, 8, 98–107 CrossRef CAS PubMed
- K. Vasilev, Z. Poh, K. Kant, J. Chan, A. Michelmore and D. Losic, Biomaterials, 2010, 31, 532–540 CrossRef CAS PubMed
- S. Zhong, R. Luo, X. Wang, L. Tang, J. Wu, J. Wang, R. Huang, H. Sun and N. Huang, Colloids Surf., B, 2014, 116, 553–560 CrossRef CAS PubMed
- S.-P. Lin, S.-Y. Huang, S.-F. Chen, L. U. Vinzons, J.-Y. Ciou and P.-J. Wong, ACS Appl. Mater. Interfaces, 2014, 6, 12071–12082 CAS
- S. Oh, K. S. Moon and S. H. Lee, J. Nanomater., 2013, 965864 Search PubMed
- M. S. Killian and P. Schmuki, Surf. Interface Anal., 2014, 46, 193–197 CrossRef CAS
- S.-J. Sun, W.-Q. Yu, Y.-L. Zhang and F.-Q. Zhang, J. Mater. Sci.: Mater. Med., 2013, 24, 1079–1091 CrossRef CAS PubMed
- C. J. Frandsen, K. S. Brammer, K. Noh, G. Johnston and S. Jin, Mater. Sci. Eng., C, 2014, 37, 332–341 CrossRef CAS PubMed
- M.-Y. Lan, S.-L. Lee, H.-H. Huang, P.-F. Chen, C.-P. Liu and S.-W. Lee, Ceram. Int., 2014, 40, 4745–4751 CrossRef CAS
- Y.-Y. Song, H. Hildebrand and P. Schmuki, Surf. Sci., 2010, 604, 346–353 CrossRef CAS
- G. G. Genchi, C. Ciofani, I. Liakos, L. Ricotti, L. Ceseracciu, A. Athanassiou, B. Mazzolai, A. Menciassi and V. Mattoli, Colloids Surf., B, 2013, 105, 144–151 CrossRef CAS PubMed
- M. M. S. Barroso, E. Freire, G. S. C. S. Limaverde, G. M. Rocha, E. J. O. Batista, G. Weissmueller, L. R. Andrade and T. Coelho-Sampaio, J. Biol. Chem., 2008, 283, 11714–11720 CrossRef PubMed
- K. Ishizaki, Y. Sugita, F. Iwasa, H. Minamikawa, T. Ueno, M. Yamada, T. Suzuki and T. Ogawa, Int. J. Nanomed., 2011, 6, 2191–2203 CAS
- T. Amna, M. S. Hassan, W.-S. Shin, H. V. Ba, H.-K. Lee, M. S. Khil and I. H. Hwang, Colloids Surf., B, 2013, 101, 424–429 CrossRef CAS PubMed
- P. Roy, S. Berger and P. Schmuki, Angew. Chem., Int. Ed., 2011, 50, 2904–2939 CrossRef CAS PubMed
- I. Paramasivam, H. Jha, N. Liu and P. Schmuki, Small, 2012, 8, 3073–3103 CrossRef CAS PubMed
- Y. R. Smith, R. S. Ray, K. Carlson, B. Sarma and M. Misra, Materials, 2013, 6, 2892–2957 CrossRef CAS
- S. Bauer, J. Park, J. Faltenbacher, S. Berger, K. von der Mark and P. Schmuki, Integr. Biol., 2009, 1, 525–532 RSC
- S. Bauer, J. Park, A. Pittrof, Y.-Y. Song, K. von der Mark and P. Schmuki, Integr. Biol., 2011, 3, 927–936 RSC
- H. Y. Seo, J.-S. Kwon, Y. R. Choi, K. M. Kim, E. H. Choi and K. N. Kim, PLoS One, 2014, 9, e113477 Search PubMed
- J. Holmberg and M. Durbeej, Cell Adhes. Migrat., 2013, 7, 111–121 CrossRef PubMed
- M. A. Lawson and P. P. Purslow, Cells Tissues Organs, 2000, 167, 130–137 CrossRef CAS PubMed
- K. von der Mark and M. Ocalan, Differentiation, 1989, 40, 150–157 CrossRef CAS PubMed
- M. F. Butler, Y. F. Ng and P. D. A. Pudney, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3941–3953 CrossRef CAS
- L. Polonchuk, J. Elbel, L. Eckert, J. Blum, E. Wintermantel and H. M. Eppenberger, Biomaterials, 2000, 21, 539–550 CrossRef CAS PubMed
- D. Wang, Y. Liu, C. Wang, F. Zhou and W. Liu, ACS Nano, 2009, 3, 1249–1257 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra00716c |
| This journal is © The Royal Society of Chemistry 2016 |