
Copolymers of ionic liquids with polymeric or metallocomplex chromophores for quasi-solid-state DSSC applications†
Abstract
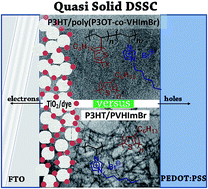
The development of copolymers based on ionic liquid vinyl monomers of the imidazole family combined with polymerizable chromophoric units is presented herein. For this end, ruthenium complexes bearing polymerizable vinyl groups or ω-end vinyl rr-poly(3-alkyl thiophene) were prepared and copolymerized with the ionic liquid monomers under free radical polymerization conditions, affording chromophore/polyelectrolyte combinations. Homopolymer ionic liquids were also synthesized to select the optimum conditions for the copolymers thereafter. All the monomers and polymers were characterized for their optical properties, and were also structurally characterized using various complementary techniques. Selected copolymers and homopolymers were tested in quasi-solid-state sensitized solar cells based on titania and regioregular poly(3-hexyl thiophene) acting as a hole-transporting semiconducting polymer. The ionic liquid, which is miscible with the hole-conductor and can be deposited with the latter, provides a functionality that, in some cases, supports an increase in open-circuit voltage, thus increasing cell efficiency.
 Please wait while we load your content...
Please wait while we load your content...

