
Exploring the low-lying structures of Aun(CO)+ (n = 1–10): adsorption and stretching frequencies of CO on various coordination sites†
Jie Wangabc,
Qing-Bo Yanb,
Jun Maa,
Xizi Caoa,
Xiaopeng Xing*a and
Xuefeng Wanga
aDepartment of Chemistry, Shanghai Key Lab of Chemical Assessment and Sustainability, Tongji University, 1239 Siping Road, Shanghai, 200092, China. E-mail: xingxp@tongji.edu.cn
bCollege of Materials Science and Opto-Electronic Technology, University of Chinese Academy of Sciences, Beijing, 100049, China
cCHINALCO Research Institute of Science and Technology, Southern District of Future Science & Technology Park, Beijing, 102209, China
First published on 11th January 2016
Abstract
The low lying structures of Aun(CO)+ (n = 1–10) within 1.0 eV from their global minima were explored using a structure searching program (Atomic Global Minimum Locator) and density functional theory (DFT) calculations. According to general chemical intuitions, CO should prefer to stay on the lowest coordination sites of gold frames. However, our calculations showed that this preference becomes very weak in large Aun(CO)+, and even negligible for their compact three dimensional structures. This character relates to the intrinsic fluxionality of gold cluster frames, which apparently readjust their bond parameters after CO adsorption. The stretching frequency of a terminal-bonded CO decreases with increasing cluster size, and structural details of the adsorption sites have insignificant influences. The bridge-bonded COs, on the contrary, has far low stretching frequencies mainly determined by structural details of the two bridge sites. The frequency variation of CO relates to the electron donation and back-donation between gold and CO. Since all the above regularities on the adsorption and frequency of CO were summarized from large amount of low lying structures rather than some specific ones, they can be used to understand adsorption of CO on active sites of real gold catalysts, whose structures are diversiform and usually unclear.
Introduction
The catalytic properties of dispersed gold species in CO oxidization reactions were discovered about two decades ago.1,2 This finding has inspired enduring efforts to explore the structures and reactivity of small gold species. Model gold species were prepared on metal oxides and characterized using various surface techniques.3–6 These experiments have shown the effects of clusters' sizes, charges, band gaps, and the defects of supporting substrates. However, the structural details of the active sites are usually unclear. Additionally, the fluxionality of small gold species has been noticed in various experimental and theoretical studies.7–10 This character could make structures of gold sites diversiform and strongly dependent on the specific surroundings. This complexity makes it even more challenging to clearly understand interactions of CO with real gold catalyst at molecular level.Naked gold clusters were generated in the gas phase and well characterized using different strategies. For example, the structures of Aun− were determined by combinations of theoretical calculations and various experiments including the ion mobility technique,11 the photoelectron spectroscopy12–14 and the trapped ion electron diffraction.15–17 The structural information of Aun+ mainly comes from ion mobility measurement and theoretical calculations.18–20 Experiments in the gas phase show that O2 tends to be an electron acceptor while CO acts as an electron donor when they interact with gold clusters.21–27 Consequently, CO prefer to stay on cationic gold sites, while O2 tend to be activated by anionic gold species. The full catalytic circles of CO oxidation were observed on some gold clusters22,28 and theoretical works proposed many possible reaction mechanisms.28–42 In spite of all those valuable clues from the gas phase studies, there are still many challenges to understand the real catalytic processes on surfaces. A significant one is that the lowest lying structures in gas phase are usually not what they are in real catalysts. An example is that Au8 on an F-center of MgO, which has a different structure43 from the lowest lying ones of the naked cationic, anionic or neutral Au8.11,18,44 In order to obtain more profound and accurate understanding on how real gold catalysts work, some general regularities applicable for various gold structures are of great meaning.
Reaction of Aun+ (n = 1–65) with CO has been studied in the gas phase. Kinetic measurements obtained the bond energies of CO on different cluster sizes.45 The subsequent work indicated that these bond energies are strongly dependent on the net positive charge around the adsorption sites.25 The infrared photo dissociation spectra of saturated Aun(CO)m+ (n = 1–10) gave the CO stretching frequencies around 2120–2180 cm−1, and additionally showed a 3D to 2D transition of the gold frames during adsorption processes.9 There have been many theoretical works on adsorptions of CO on cationic gold species.9,25,36,41,42,45–48 Most of these studies focused on predicting the global minima which possibly present in the gas phase experiments. The CO adsorptions on supported cationic gold species also attracted great attentions. The IR bands around 2150 cm−1 in surface studies have been empirically assigned to those on cationic gold sites,4,49,50 which is slightly higher than those of COs on small neutral gold clusters (2050–2150 cm−1).51
In this work, we explored the low lying structures of [Aun(CO)]+ (n = 1–10) using a structure searching program (Atomic Global Minimum Locator) and density functional theory (DFT) calculations. Other than making great efforts to determine the global minimum of each size, we considered the characters and properties of a large amount of low lying structures within 1.0 eV energy range. This energy value is roughly the order of interacting energies between substrates and small metal species. Our results revealed some general regularities about CO adsorption and stretching frequency on different coordination sites of gold. The obtained conclusions should be useful to understand what happens on the active gold sites in real catalysts.
Calculation methods
All calculations were performed using the GAUSSIAN 09 suite package52 and a structure searching program of Atomic Global Minimum Locator.53 This searching program has been used to locate the lowest structures of metal clusters and organic complexes.54–56 Firstly, we combined the simulated annealing algorithm method in Atomic Global Minimum Locator and the DFT method based on the hybrid Becke–Lee–Yang–Parr functional (B3LYP)57,58 in GAUSSIAN 09 to locate the low lying structures of [Aun(CO)]+ (n = 1–10). The B3LYP method has been shown to be reliable in predicting the structures of small gold species, evaluating the relative energies of different isomers, and calculating the stretching frequencies of CO adsorbed on gold.25,47,59–65 In the structural searching processes, the lanl2dz basis set was used for Au and the 6-31G(d) basis set for C and O (level I). As supplementary to this first structural searching strategy, we manually designed structures by putting CO on each of the possible adsorption sites of all previously reported structures of Aun+/0/−,11,12,18,19,66 and also optimized them at aforementioned level I. After removing the duplicated ones from these two structural locating strategies, the frequencies of all structural candidates were analyzed to make sure they are real minima. For Aun(CO)+ (n = 1–5), the optimized structures from level I were further optimized using B3LYP method with larger basis sets, Aug-cc-PVTZ-PP for Au67,68 and Aug-cc-PVTZ for C and O (level II).69,70 The CO frequencies were calculated using analytic second derivatives at both level I and level II. For Aun(CO)+ (n = 6–10), the single point energies of the optimized structures at level I were recalculated at level II and the CO frequencies were only calculated at level I.Results and discussion
A. Large quantities of low lying structures of Aun(CO)+
For Aun(CO)+ (n = 1–5), the optimization at both level I and level II got consistent structures and roughly same relative energies. The low lying structures from these two levels were displayed in Fig. 1, which are sequenced according to the relative energies at level II. The determined global minima for AunCO+ (n = 1–6) are consistent with those reported in ref. 45 and 48. The second lowest lying structures for both Au2CO+ and Au3CO+ contain a bridge-bonded CO, which are 0.6–0.8 eV and 1.5–1.6 eV higher than their global minima, respectively. For Au4(CO)+, the structures 2–5 are within 1.0 eV from its lowest lying structure 1, and the structure 5 contains a bridge-bonded CO. For Au5(CO)+, the structures 1–4 are within the 1.0 eV energy range, and their energy sequence is consistent with that in ref. 45.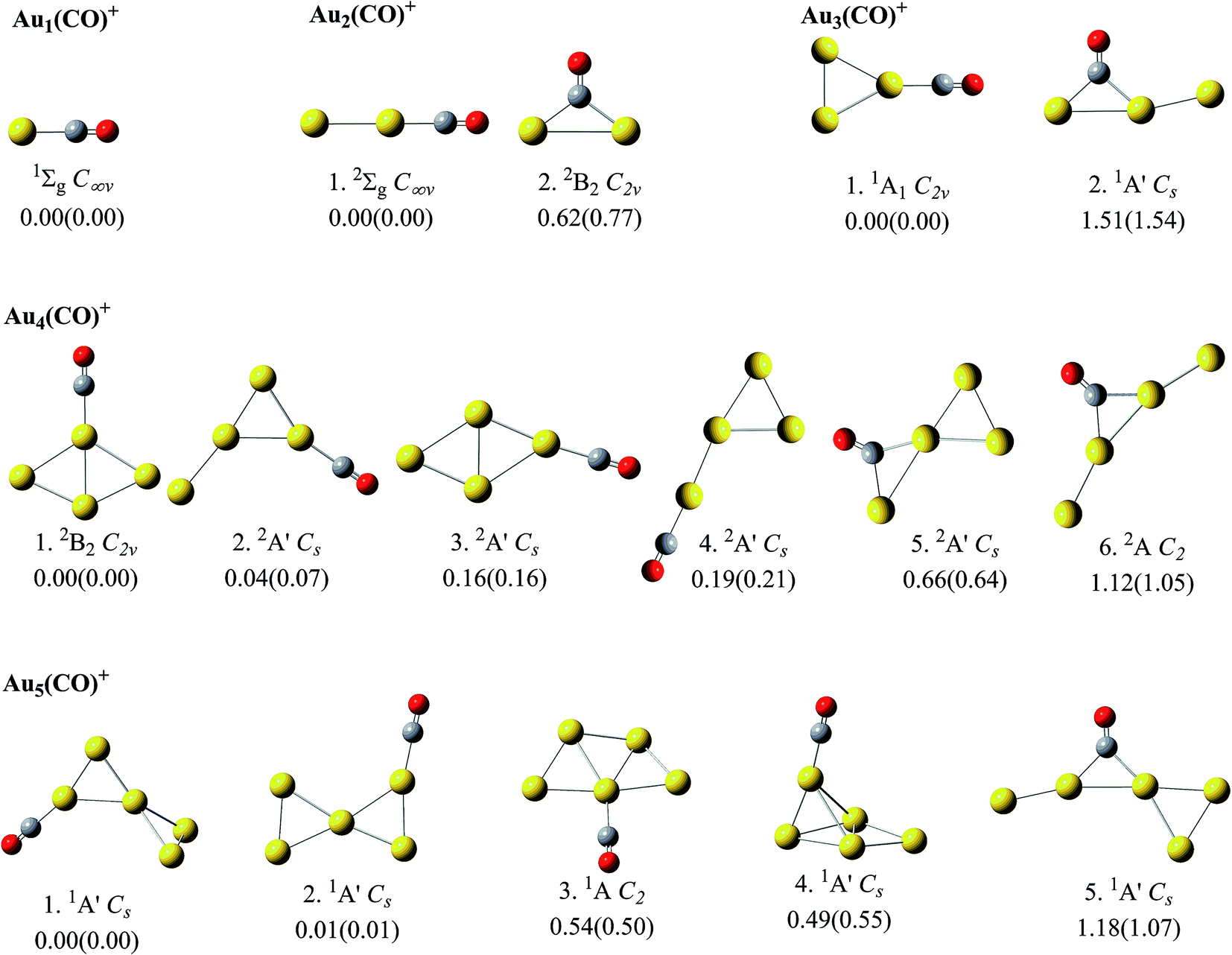 | ||
| Fig. 1 The theoretical low lying structures of Aun(CO)+ (n = 1–5). Their electronic states and geometries are indicated. The numbers inside and outside of the parentheses indicate the relative energies from level I and level II, respectively. Level I: B3LYP method with lanl2dz basis set for Au and 6-31G(d) basis sets for C and O. Level II: B3LYP method with Aug-cc-PVTZ-PP basis set for Au and Aug-cc-PVTZ basis sets for C and O. | ||
The isomer populations of Aun(CO)+ increase enormously from n = 6. Since the results of AunCO+ (n = 1–5) indicate that further optimization at level II does not apparently change the structures from level I, Aun(CO)+ (n = 6–10) were only optimized using level I and only the single point energies were recalculated at level II. All the structures of Aun(CO)+ (n = 6–10) within 1.0 eV energy range were summarized in ESI Fig. S1–S5.† They were grouped as terminal adsorption complexes and bridge adsorption ones. Each group was sequenced separately according to the relative energies from level II. In Fig. 2, we further classified the low lying structures of Aun(CO)+ (n = 6–10) according to structural details of their adsorption sites. The nomenclature and the symbols of each type were shown in Fig. 2a. The 2D-II, 2D-III and 2D-IV means that the adsorption happens on a two dimensional gold site, in which the gold atom bonding CO connects with 2, 3 and 4 other gold atoms, respectively. Similarly, the 3D-III, 3D-IV, 3D-V and 3D-VI means that the adsorption gold site is three dimensional and the gold atom bonding CO connects with 3, 4, 5 and 6 other gold atoms, respectively. The bridge symbol means CO bridges on two gold atoms. The geometries of the three lowest lying ones for each type (if they are within the 1.0 eV energy range) were listed inside the figure panels. For Au6(CO)+, twenty low lying structures were shown in Fig. S1† and 2b. The structures 1–3 correspond to the three lowest lying ones predicted in ref. 45, while their relative energies are slightly different. The forth structure of Au6(CO)+ reported in ref. 45 is not a real minimum at two theoretical levels in this work. There are fourteen low lying structures of Au7(CO)+ in Fig. S2† and 2c. The structures 1–4 correspond to the four lowest lying ones in ref. 45, and there are only a small differences on the structural details and energy intervals. For Au8(CO)+, forty seven isomers were located and shown in Fig. S3† and 2d. The three lowest lying ones 1–3 are same as those reported in ref. 45, while the forth lowest lying structure of Au8(CO)+ in ref. 45 corresponds to the structure 11 of this work. The two lowest lying structures 1 and 2 of Au8(CO)+ are predicted to be planar even that of naked Au8+ were determined to be three dimensional. For Au9(CO)+, there are thirty two structures listed in Fig. S4† and 2e. Structures 1 and 4 correspond to the lowest lying Au9+ structure with CO adsorbed on different sites. For Au10(CO)+, there are totally eighty five structures displayed in Fig. S5† and 2f. Structures 1–5 are within 0.1 eV and the ones 1–3 correspond to the lowest lying Au10+ structure with CO on different adsorption sites.
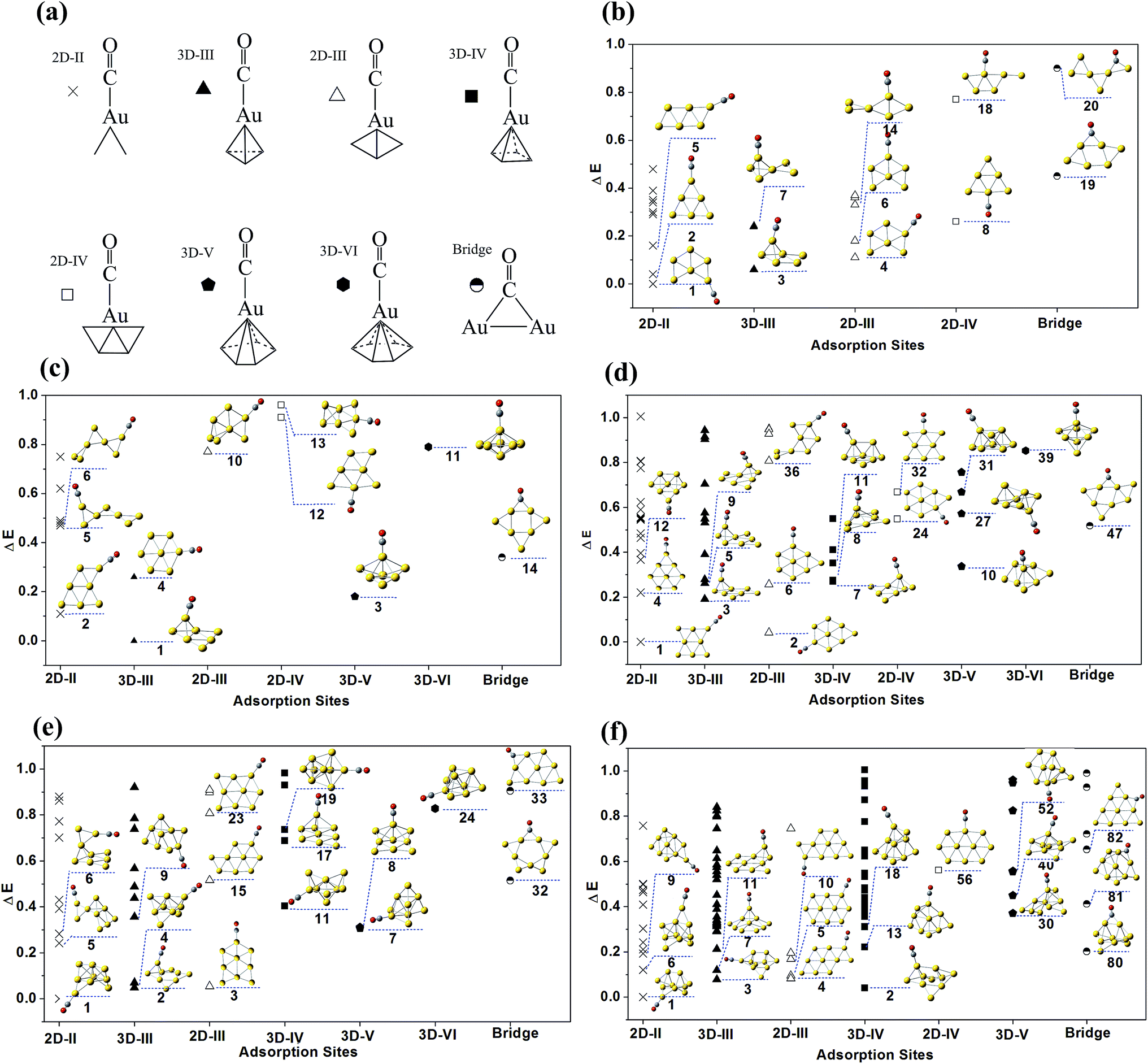 | ||
| Fig. 2 The theoretical low lying structures of Aun(CO)+ (n = 6–10). The structures were optimized at level I and their relative energies were recalculated at level II. All the structures are classified according to the geometries of CO adsorption sites. The classification and the nomenclature are shown in (a). The results of Aun(CO)+ (n = 6–10) are shown in (b)–(f), respectively. In each panel of (b)–(f), the horizontal axis stands for different types of adsorption sites and the vertical axis stands for the relative energy. The three lowest structures for each type of adsorption site (if there are) are displayed. The structures of each size are sequenced and numbered according to their relative energies at level II, which were included in ESI Fig. S1–S5.† | ||
B. A weak preference of CO on the lowest coordination sites
According to Fig. 1 and 2, most of the global minima of Aun(CO)+ have the CO on their lowest coordination sites, which means the terminal gold atom for n = 1 and 2 and the 2D-II type (shown in Fig. 2a) for n = 3, 5, 6, and 8–10. This is consistent with the general chemical intuitions that CO tend to be adsorbed on the lowest coordination site of metals. However, when we considered all low lying isomers of Aun(CO)+ within 1.0 eV from the global minimum, we found that the ratio of the structures with CO on high coordination sites increases noticeably with the increase of cluster sizes. For Au10(CO)+, the isomer populations belonging to 2D-II, 3D-III and 3D-IV types are nearly equal. Even the bridge type of Au10(CO)+ have 6 isomers and the lowest lying one is only 0.2 eV higher than the global minimum. The energy levels are quite close for the low-lying structures of Aun(CO)+ (n = 1–10) in this report and those of Aun(CO)+ (n = 1–6) in previous reports45,48 with CO on different coordination sites. According to this observation, we can say there is indeed a preference of CO on the lowest coordination site on small gold clusters, however this preference becomes weak when cluster size increases. We found this argument is especially true for the compact three dimensional structures, as shown by the representative examples in Fig. 3. The three structures of Au8(CO)+ (11, 12 and 13 from Fig. S3†) correspond to the lowest lying 3D structure of Au8+ (ref. 18) with CO on the different sites, and their energy differences are within 0.05 eV. Similarly, the structures of Au9(CO)+ (1 and 4 from Fig. S4†) correspond the global minimum of Au9+ (ref. 18) with CO on its different sites, and their energy difference is 0.07 eV; the three structures of Au10(CO)+ (1, 2 and 3 from Fig. S5†) correspond to CO adsorption on different sites of the global minimum of Au10+,18 and their energy differences are within 0.08 eV. In a previous theoretical studies, CO was put on all possible sites of Au21+ and the bond energies are shown dramatically different.25 The physical origin of that large differences were attributed to different positive charges on various adsorption sites. We noticed that the gold frame was fixed in that calculation, therefore the results show a clear relation between the bonding energies and the positive charge of various sites. In this work, every structures were fully relaxed and optimized after CO was added, and we saw apparent readjustment of the bond parameters of the gold frames. Combining the figures revealed in ref. 25 and this work, we can say that the net positive charges on individual cite indeed play an important role for adsorbing CO, while a 3D gold cluster can adjust its frame, arriving structures with nearly same energies even the CO is initially put on its different sites. This character can be attributed to the intrinsic fluxionality of gold, which has been considered as one of the physical origins for their catalytic properties.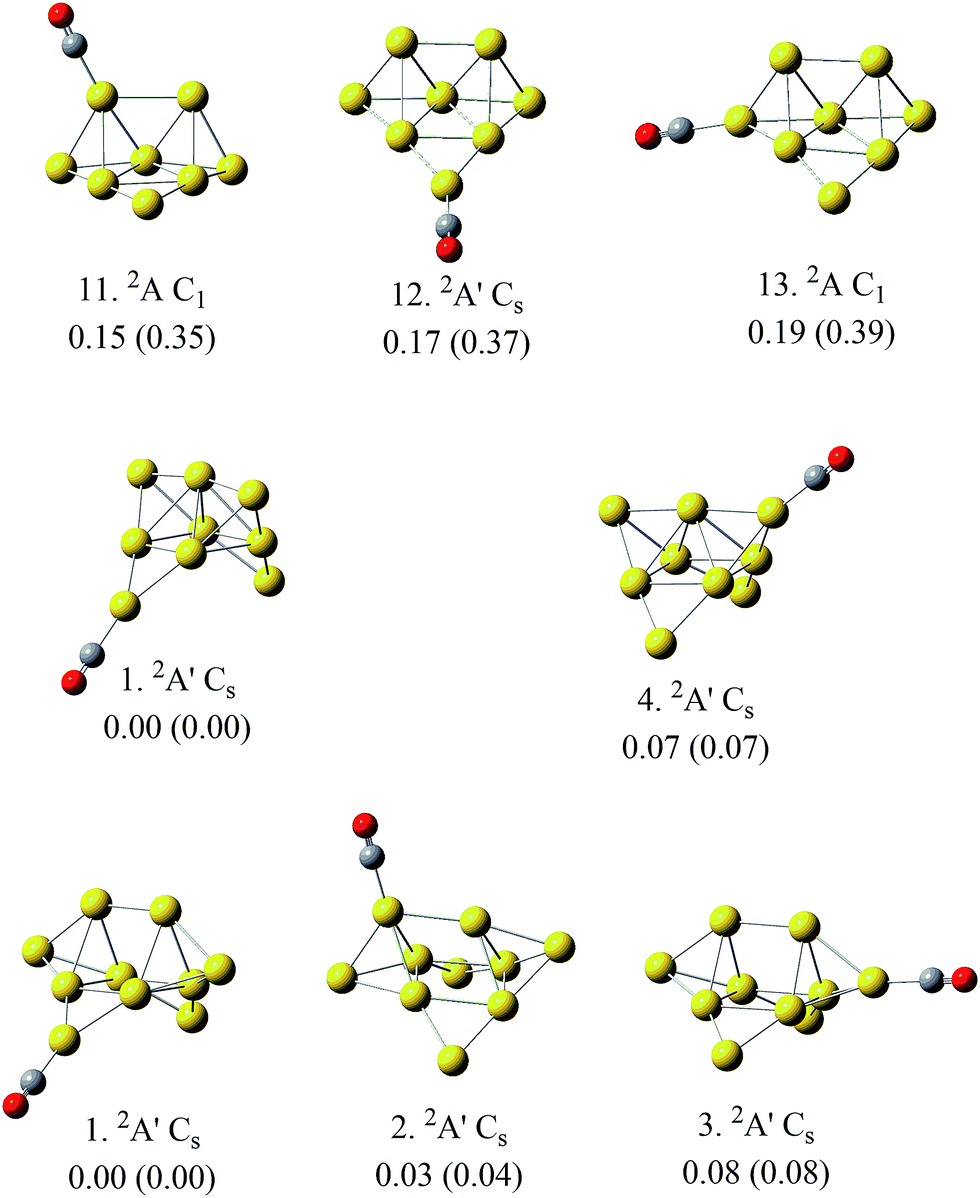 | ||
| Fig. 3 The representative structures of Aun(CO)+ (n = 8–10), in which the gold moieties are compact three dimensional with CO on various adsorption sites. Their electronic states and geometries are indicated. The numbers inside and outside of the parentheses indicate their energies relative to the global minimum from level I and level II, respectively. The series numbers for these structures come from ESI Fig. S3–S5.† | ||
C. Calculating the stretching frequencies of COs in various structures
We calculated the CO stretching frequencies of all structures of Aun(CO)+ (n = 1–5) in Fig. 1 using both level I and level II theories. Two scaling factors, 0.9703 and 0.9710, were obtained when dividing the experimental frequency (2143 cm−1) of a free CO molecule by the two calculated ones, 2208.6 cm−1 and 2207.1 cm−1, at level I and level II, respectively. We also tested these two theoretical levels by calculating the CO frequency in Au3(CO)3+, and got the scaled frequencies of 2187 cm−1 and 2178 cm−1, respectively. Both of them are reasonably consistent with the experiment one (2182 cm−1) in ref. 9. Table 1 lists the calculated CO stretching frequencies in Aun(CO)+ (n = 1–5). Those of the terminal-bonded CO at level I are slightly higher than those at level II. The differences are within 15 cm−1. The values of AuCO+ in Table 1 (2237 cm−1 and 2234 cm−1) are also very close to that (∼2250 cm−1) predicted using CCSD methods.71 For most of the bridge-bonded COs, the stretching frequencies from the two levels are also very close. The only exception is the structure 5 of Au4(CO)+ (see Fig. 1 and Table 1). The difference between CO frequencies from these two levels is about 50 cm−1. However, the frequency sequences from two levels are always consistent. For Aun(CO)+ (n = 6–10), we only used level I calculating their CO stretching frequencies. In order to make a systematical comparison, Fig. 4 lists all CO frequencies of Aun(CO)+ (n = 1–10) from level I. The structures were classified according to the type of the adsorption sites, same as what has been done in Fig. 2. The new dash symbol ‘-’ for Aun(CO)+ (n = 1, 2 and 4) stands for the structures where CO bonds on a terminal gold atom.| Structures | CO stretching frequencies (cm−1) | ||
|---|---|---|---|
| Level I | Level II | ||
| Au(CO)+ | 2237 | 2234 | |
| Au2(CO)+ | 1 | 2215 | 2206 |
| 2 | 1918 | 1935 | |
| Au3(CO)+ | 1 | 2198 | 2189 |
| 2 | 1994 | 2000 | |
| Au4(CO)+ | 1 | 2186 | 2173 |
| 2 | 2188 | 2174 | |
| 3 | 2193 | 2181 | |
| 4 | 2190 | 2179 | |
| 5 | 1948 | 2000 | |
| 6 | 2025 | 2019 | |
| Au5(CO)+ | 1 | 2183 | 2169 |
| 2 | 2183 | 2170 | |
| 3 | 2171 | 2160 | |
| 4 | 2188 | 2176 | |
| 5 | 2018 | 2010 | |
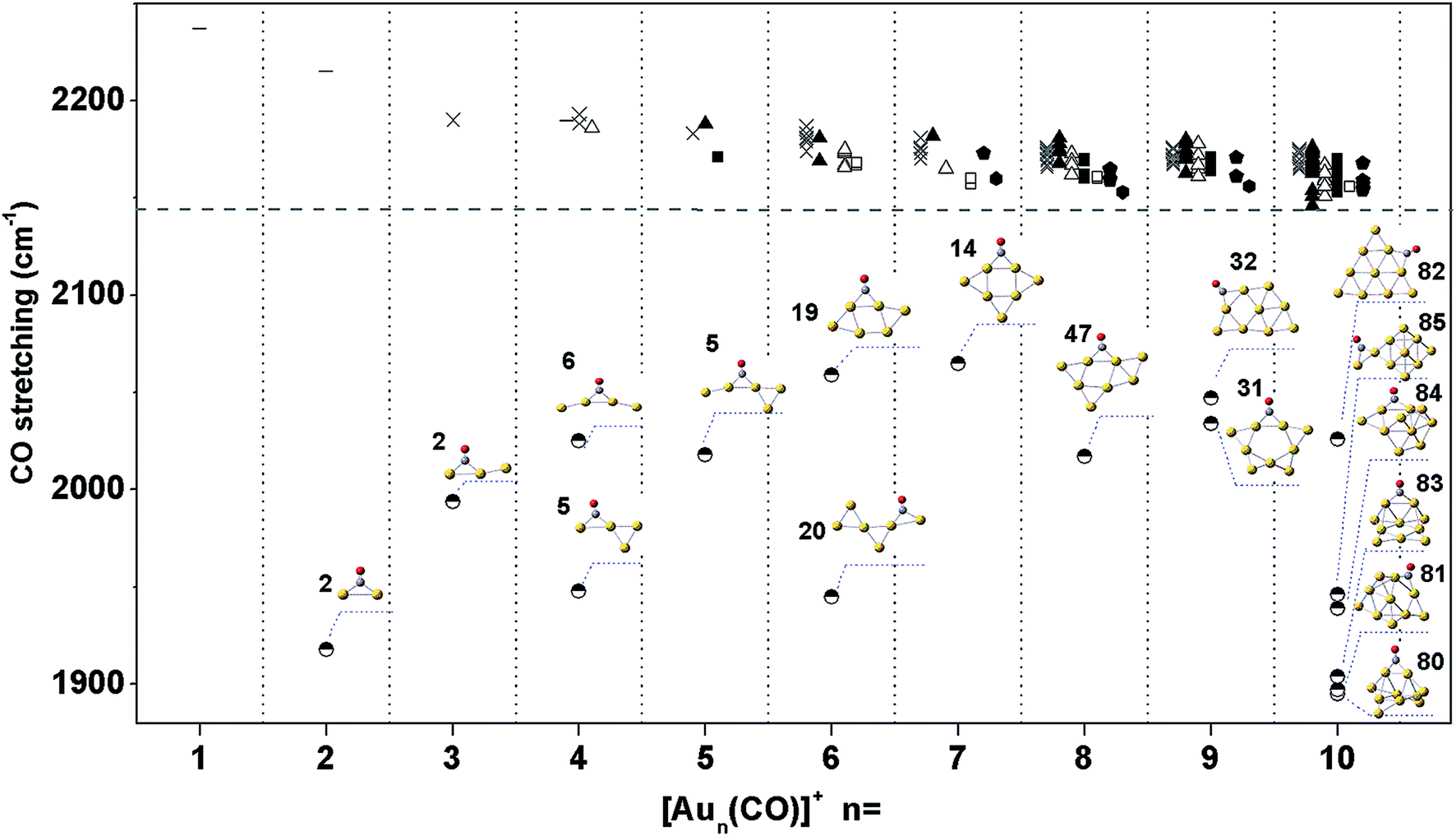 | ||
| Fig. 4 The stretching frequencies of CO in low lying structures of Aun(CO)+ (n = 1–10) (at level I). The classification and the nomenclature of these structure are same as that in Fig. 2(a). The dash symbol ‘-’ indicates CO on a terminal gold atom. The horizontal dashed line indicates the stretching frequency of a free CO molecule. The structures with a bridge-bonded CO are displayed and their series numbers come from ESI Fig. S1–S5.† | ||
D. Dependence of CO stretching frequencies on the adsorption sites
In Fig. 4, the frequencies of all terminal-bonded COs are higher than that of a free CO. This blue shift has been observed in various cationic gold carbonyls, such as Aun(CO)m+ in the gas phase and Auδ+ on oxide surfaces.4,9,49,71,72 The σ orbital of CO has partially anti-bonding characters, and the polarization of this orbital toward cationic gold species leads to an enhancement of the C–O bonding, and therefore the blue shift of the CO stretching frequency. With increasing cluster size, the positive charge is diluted and the pulling of CO σ orbital by gold sites decreases, making the blue shift decreases. For various isomers of Aun(CO)+ (n = 4–8), there is an unapparent trend that the CO frequency decreases with increasing coordination number of the adsorption site, and this trend nearly disappears for Au9,10(CO)+. Generally, bridge-bonded COs have lower frequencies than those of terminal-bond ones.51 In Fig. 4, the frequencies of bridge-bonded COs expand from lower than 1900 cm−1 to around 2100 cm−1. These values seem independent on the cluster sizes, while strongly dependent on the structural details of the bridge sites. For example, the CO bridging on the planar structures in Au4(CO)+-6, Au5(CO)+-5, Au6(CO)+-19, Au7(CO)+-14, Au8(CO)+-47, Au9(CO)+-31, Au9(CO)+-32 or Au10(CO)+-82 has frequencies around 2050 cm−1; the CO bridging on a terminal gold atom and another one belonging to any other type, like that in Au2(CO)+-2, Au3(CO)+-2, Au4(CO)+-5, Au6(CO)+-20 or Au10(CO)+-85 has frequencies around 1950 cm−1; the CO bridging on three dimensional sites of gold has frequencies around 1900 cm−1, like those in Au10(CO)+-80, 81 and 83. The diluting of positive charges along with increasing cluster size does not have significant effects on the frequencies of these bridge-bonded COs.Usually, the stretching frequencies of adsorbed COs are closely related to the electron donation to and back-donation from metal moieties. We analyzed the frontier molecular orbitals in three representative structures Au10(CO)+-80, 82 and 1, which have a bridge CO on 3D sites (Fig. 5a), a bridge CO on 2D sites (Fig. 5b) and a terminal-bonded CO on a 2D-II site (Fig. 5c), respectively. The HOMO orbitals in Fig. 5a and b indicate electron back donation from the gold moieties to the π* of CO, and therefore lead to red-shifts of the CO stretching frequency. Qualitatively, the back donation in the structure of Fig. 5a is more significant than that of Fig. 5b, which leads to the lowest vibration frequency of CO bridging on 3D sites. The HOMO in Fig. 5c comes mainly from gold moiety and no back donation exists in this case. The HOMO−1 in Fig. 5c correspond to polarization of the σ orbital of CO (partially anti-bonding) toward the gold, causing the apparent blue-shift relative to that of a free CO.
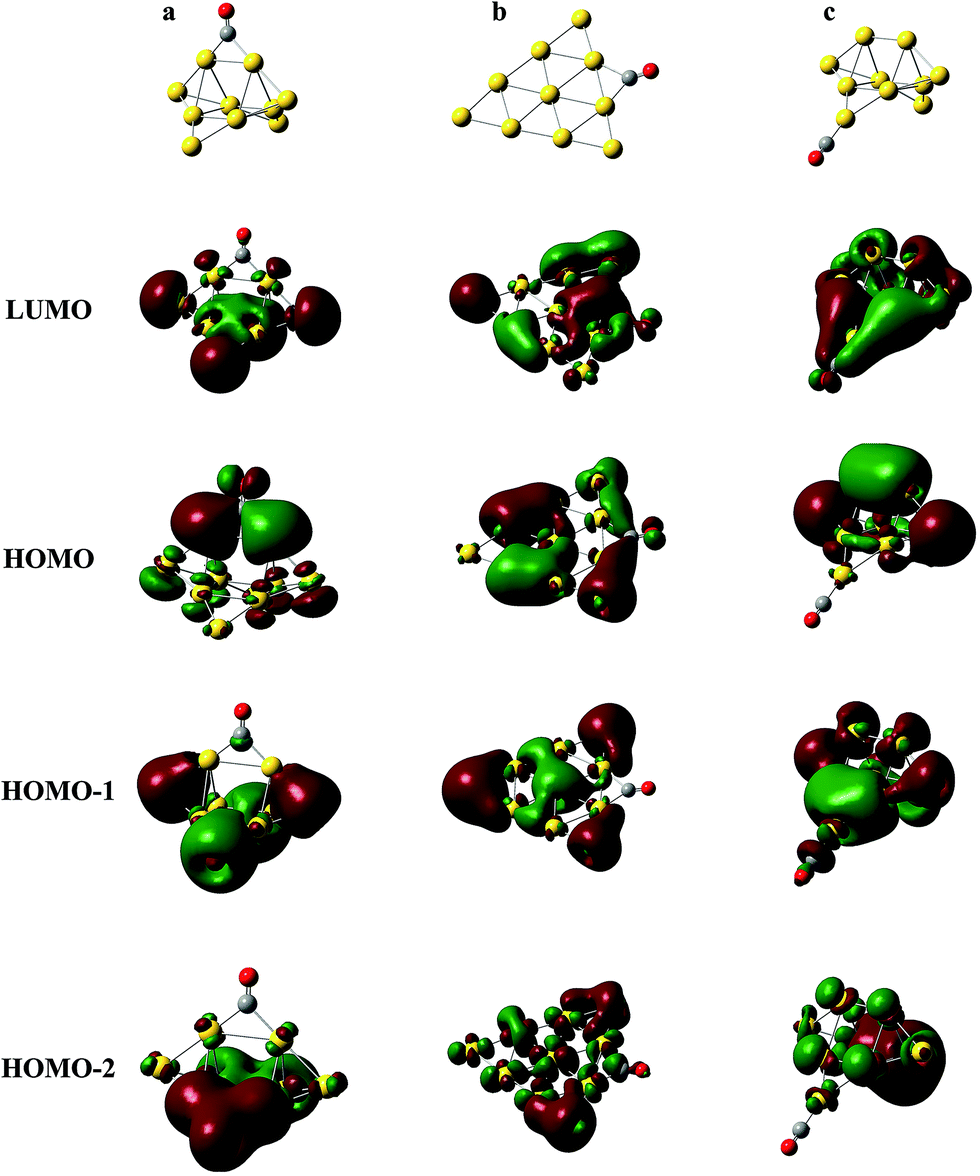 | ||
| Fig. 5 The frontier molecular orbitals (at level I) of three representative structures of Au10(CO)+, which have a bridge-bonded CO on two 3D sites (a), a bridge-bonded CO on two 2D sites (b) and a terminal-bonded CO on the lowest coordination site (c), respectively. | ||
Conclusions
We systematically searched the low lying structures of Aun(CO)+ (n = 1–10) using Atomic Global Minimum Locator and DFT calculations. Using the large amount of structures as model systems, we summarized some general regularities on CO adsorption and stretching frequencies on various coordination sites of gold species. Firstly, the preference of CO staying on the lowest coordination sites decreases with increasing cluster size. For the compact 3D gold frames of Au8+, Au9+, and Au10+, the structures with CO on different coordination sites have nearly degenerated energies. The frequencies of terminal-bonded COs decrease with increasing cluster sizes and diluting of positive charge. There is a very weak dependence of these CO frequencies on the structural details. Those of bridge-bonded COs, on the contrary, are mainly determined by the structures of their two bridging sites. Those bridging on 3D gold sites have apparently lower frequencies than those on 2D or dangling gold atoms. The weak preference of CO on certain coordination sites and the variation trend of CO stretching frequencies were well rationalized using the intrinsic fluxionality of gold and the electron transfer mechanism, respectively. Usually, the structural details of real catalysts are diversiform and unknown. The above regularities from large amount of various gold structures should be applicable for understanding adsorption of COs on cationic gold sites in real catalysts.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21103226 and 21273278), the Fundamental Research Funds for the Central Universities, the Ministry of Science and Technology of China (Grant No. 2012YQ22011307), and Science & Technology Commission of Shanghai Municipality (14DZ2261100). The calculations were carried out on the Deepcomp7000 of Supercomputing Center, Computer Network Information Center of Chinese Academy of Sciences, and the computing server in Department of Chemistry, Tongji University.Notes and references
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 405–408 CrossRef CAS.
- M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301–309 CrossRef CAS.
- M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 1647–1650 CrossRef CAS PubMed.
- Y. Hao, M. Mihaylov, E. Ivanova, K. Hadjiivanov, H. Knozinger and B. C. Gates, J. Catal., 2009, 261, 137–149 CrossRef CAS.
- J. C. Fierro-Gonzalez, J. Guzman and B. C. Gates, Top. Catal., 2007, 44, 103–114 CrossRef CAS.
- A. Sanchez, S. Abbet, U. Heiz, W. D. Schneider, H. Hakkinen, R. N. Barnett and U. Landman, J. Phys. Chem. A, 1999, 103, 9573–9578 CrossRef CAS.
- H. Hakkinen, W. Abbet, A. Sanchez, U. Heiz and U. Landman, Angew. Chem., Int. Ed., 2003, 42, 1297–1300 CrossRef CAS PubMed.
- H.-J. Zhai, L.-L. Pan, B. Dai, B. Kiran, J. Li and L.-S. Wang, J. Phys. Chem. C, 2008, 112, 11920–11928 CAS.
- A. Fielicke, G. von Helden, G. Meijer, D. B. Pedersen, B. Simard and D. M. Rayner, J. Am. Chem. Soc., 2005, 127, 8416–8423 CrossRef CAS PubMed.
- X.-F. Yang, Y.-L. Wang, Y.-F. Zhao, A.-Q. Wang, T. Zhang and J. Li, Phys. Chem. Chem. Phys., 2010, 12, 3038–3043 RSC.
- F. Furche, R. Ahlrichs, P. Weis, C. Jacob, S. Gilb, T. Bierweiler and M. M. Kappes, J. Chem. Phys., 2002, 117, 6982–6990 CrossRef CAS.
- H. Hakkinen, B. Yoon, U. Landman, X. Li, H. J. Zhai and L. S. Wang, J. Phys. Chem. A, 2003, 107, 6168–6175 CrossRef.
- S. Bulusu, X. Li, L. S. Wang and X. C. Zeng, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8326–8330 CrossRef CAS PubMed.
- B. Yoon, P. Koskinen, B. Huber, O. Kostko, B. von Issendorff, H. Hakkinen, M. Moseler and U. Landman, ChemPhysChem, 2007, 8, 157–161 CrossRef CAS PubMed.
- X. P. Xing, B. Yoon, U. Landman and J. H. Parks, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 165423 CrossRef.
- A. Lechtken, C. Neiss, M. M. Kappes and D. Schooss, Phys. Chem. Chem. Phys., 2009, 11, 4344–4350 RSC.
- M. P. Johansson, A. Lechtken, D. Schooss, M. M. Kappes and F. Furche, Phys. Rev. A, 2008, 77, 053202 CrossRef.
- S. Gilb, P. Weis, F. Furche, R. Ahlrichs and M. M. Kappes, J. Chem. Phys., 2002, 116, 4094–4101 CrossRef CAS.
- P. Weis, T. Bierweiler, E. Vollmer and M. M. Kappes, J. Chem. Phys., 2002, 117, 9293–9297 CrossRef CAS.
- J. R. Soto, B. Molina and J. J. Castro, RSC Adv., 2014, 4, 8157–8164 RSC.
- B. E. Salisbury, W. T. Wallace and R. L. Whetten, Chem. Phys., 2000, 262, 131–141 CrossRef CAS.
- W. T. Wallace and R. L. Whetten, J. Am. Chem. Soc., 2002, 124, 7499–7505 CrossRef CAS PubMed.
- D. W. Yuan and Z. Zeng, J. Chem. Phys., 2004, 120, 6574–6584 CrossRef CAS PubMed.
- W. Huang, H. J. Zhai and L. S. Wang, J. Am. Chem. Soc., 2010, 132, 4344–4351 CrossRef CAS PubMed.
- M. Neumaier, F. Weigend, O. Hampe and M. M. Kappes, Faraday Discuss., 2008, 138, 393–406 RSC.
- T. M. Bernhardt, J. Hagen, S. M. Lang, D. M. Popolan, L. D. Socaciu-Siebert and L. Woste, J. Phys. Chem. A, 2009, 113, 2724–2733 CrossRef CAS PubMed.
- J. Hagen, L. D. Socaciu, M. Elijazyfer, U. Heiz, T. M. Bernhardt and L. Woste, Phys. Chem. Chem. Phys., 2002, 4, 1707–1709 RSC.
- L. D. Socaciu, J. Hagen, T. M. Bernhardt, L. Woste, U. Heiz, H. Hakkinen and U. Landman, J. Am. Chem. Soc., 2003, 125, 10437–10445 CrossRef CAS PubMed.
- H. Hakkinen and U. Landman, J. Am. Chem. Soc., 2001, 123, 9704–9705 CrossRef CAS PubMed.
- T. M. Bernhardt, Int. J. Mass Spectrom., 2005, 243, 1–29 CrossRef CAS.
- H. C. Fang, Z. H. Li and K. N. Fan, Phys. Chem. Chem. Phys., 2011, 13, 13358–13369 RSC.
- Z. Yuan, X.-N. Li and S.-G. He, J. Phys. Chem. Lett., 2014, 5, 1585–1590 CrossRef CAS PubMed.
- X.-N. Li, Z. Yuan and S.-G. He, J. Am. Chem. Soc., 2014, 136, 3617–3623 CrossRef CAS PubMed.
- Z.-Y. Li, Z. Yuan, X.-N. Li, Y.-X. Zhao and S.-G. He, J. Am. Chem. Soc., 2014, 136, 14307–14313 CrossRef CAS PubMed.
- F. Wang, D. J. Zhang, X. H. Xu and Y. Ding, J. Phys. Chem. C, 2009, 113, 18032–18039 CAS.
- A. Prestianni, A. Martorana, F. Labat, I. Ciofini and C. Adamo, J. Mol. Struct.: THEOCHEM, 2009, 903, 34–40 CrossRef CAS.
- G. E. Johnson, R. Mitric, V. Bonacic-Koutecky and A. W. Castleman, Chem. Phys. Lett., 2009, 475, 1–9 CrossRef CAS.
- M. Boronat and A. Corma, Dalton Trans., 2010, 39, 8538–8546 RSC.
- R. C. Deka, D. Bhattacharjee, A. K. Chakrabartty and B. K. Mishra, RSC Adv., 2014, 4, 5399–5404 RSC.
- Y. Gao, N. Shao, S. Bulusu and X. C. Zeng, J. Phys. Chem. C, 2008, 112, 8234–8238 CAS.
- A. Prestianni, A. Martorana, I. Ciofini, F. Labat and C. Adamo, J. Phys. Chem. C, 2008, 112, 18061–18066 CAS.
- A. Prestianni, A. Martorana, F. Labat, I. Ciofini and C. Adamo, J. Phys. Chem. B, 2006, 110, 12240–12248 CrossRef CAS PubMed.
- B. Yoon, H. Hakkinen, U. Landman, A. S. Worz, J. M. Antonietti, S. Abbet, K. Judai and U. Heiz, Science, 2005, 307, 403–407 CrossRef CAS PubMed.
- S. A. Serapian, M. J. Bearpark and F. Bresme, Nanoscale, 2013, 5, 6445–6457 RSC.
- M. Neumaier, F. Weigend, O. Hampe and M. M. Kappes, J. Chem. Phys., 2005, 122, 104702 CrossRef PubMed.
- D. M. Popolan, M. Nossler, R. Mitric, T. M. Bernhardt and V. Bonacic-Koutecky, Phys. Chem. Chem. Phys., 2010, 12, 7865–7873 RSC.
- P. Schwerdtfeger, M. Lein, R. P. Krawczyk and C. R. Jacob, J. Chem. Phys., 2008, 128, 124302 CrossRef PubMed.
- X. Wu, L. Senapati, S. K. Nayak, A. Selloni and M. Hajaligol, J. Chem. Phys., 2002, 117, 4010–4015 CrossRef CAS.
- A. S. Worz, U. Heiz, F. Cinquini and G. Pacchioni, J. Phys. Chem. B, 2005, 109, 18418–18426 CrossRef PubMed.
- F. Menegazzo, M. Manzoli, A. Chiorino, F. Boccuzzi, T. Tabakova, M. Signoretto, F. Pinna and N. Pernicone, J. Catal., 2006, 237, 431–434 CrossRef CAS.
- L. Jiang and Q. Xu, J. Phys. Chem. A, 2005, 109, 1026–1032 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegeland et al., Gaussian 09, Revision A02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- http://www.sourceforge.net/projects/atomicglobalmin/.
- B. B. Averkiev, S. Call, A. I. Boldyrev, L.-M. Wang, W. Huang and L.-S. Wang, J. Phys. Chem. A, 2008, 112, 1873–1879 CrossRef CAS PubMed.
- S. T. Call, D. Y. Zubarev and A. I. Boldyrev, J. Comput. Chem., 2007, 28, 1177–1186 CrossRef CAS PubMed.
- J. Clark, S. T. Call, D. E. Austin and J. C. Hansen, J. Phys. Chem. A, 2010, 114, 6534–6541 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- M. Neumaier, F. Weigend, O. Hampe and M. M. Kappes, J. Chem. Phys., 2006, 125, 104308 CrossRef PubMed.
- Q. Xu and L. Jiang, J. Phys. Chem. A, 2006, 110, 2655–2662 CrossRef CAS PubMed.
- A. Martinez, J. Phys. Chem. C, 2010, 114, 21240–21246 CAS.
- H.-C. Fang, Z. H. Li and K.-N. Fan, Phys. Chem. Chem. Phys., 2011, 13, 13358–13369 RSC.
- P. V. Nhat, T. B. Tai and M. T. Nguyen, J. Chem. Phys., 2012, 137, 164312 CrossRef PubMed.
- S. Rai, N. V. S. Kumar and H. Singh, Bull. Mater. Sci., 2012, 35, 291–295 CrossRef CAS.
- Z. Jamshidi, H. Farhangian and Z. A. Tehrani, Int. J. Quantum Chem., 2013, 113, 1062–1070 CrossRef CAS.
- H. Hakkinen and U. Landman, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 2287–2290 CrossRef.
- K. A. Peterson and C. Puzzarini, Theor. Chem. Acc., 2005, 114, 283–296 CrossRef CAS.
- D. Figgen, G. Rauhut, M. Dolg and H. Stoll, Chem. Phys., 2005, 311, 227–244 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- J. Velasquez, B. Njegic, M. S. Gordon and M. A. Duncan, J. Phys. Chem. A, 2008, 112, 1907–1913 CrossRef CAS PubMed.
- A. Deka, R. C. Deka and A. Choudhury, Chem. Phys. Lett., 2010, 490, 184–188 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: All theoretical structures of Aun(CO)+ (n = 6–10) within 1.0 eV energy range were summarized in ESI Fig. S1–S5. See DOI: 10.1039/c5ra25494a |
| This journal is © The Royal Society of Chemistry 2016 |