
Synthesis of 1,2,4-triazine derivatives via [4 + 2] domino annulation reactions in one pot†
Dong Tangac,
Jing Wangac,
Ping Wuac,
Xin Guoac,
Ji-Hui Lib,
Sen Yangac and
Bao-Hua Chen*ac
aState Key Laboratory of Applied Organic Chemistry, Lanzhou University, Lanzhou, Gansu 730000, China
bCollege of Materials and Chemical Engineering, Hainan University, Haikou, 570228, China
cKey Laboratory of Nonferrous Metal Chemistry and Resources Utilization of Gansu Province, Lanzhou 730000, China. E-mail: chbh@lzu.edu.cn; Fax: +86-931-891-2582
First published on 21st January 2016
Abstract
Simple and efficient [4 + 2] domino annulation reactions have been developed for the synthesis of 1,2,4-triazine derivatives. The strategies exhibit high performance with moderate to high yields, using easily available materials including ketones, aldehydes, alkynes, secondary alcohols and alkenes, representing a powerful tool for the formation of potentially biologically active derivatives.
Introduction
The 1,2,4-triazine moiety represents an important class of heterocycles, and 1,2,4-triazine derivatives have been reported to possess a broad spectrum of biological activities, including anti-convulsant,1a anti-cancer,1b anti-cytokine,1c anti-tumor,1d anti-viral,1e anti-hypertensive1f and anti-malarial1g activities. In addition, some important metal coordinating ligands contain the 1,2,4-triazine skeleton.2 These classes of compounds are significant and useful; however, there is a shortage of systemic, operable and versatile strategies to construct 1,2,4-triazine skeletons to meet the demand of synthetic chemistry. Among the known methods for the synthesis of 1,2,4-triazines, the reactions of amidrazones and 1,2-diketone compounds are the most available protocols to form 3,5-disubstituted or 3,5,6-trisubstituted 1,2,4-triazines.3 Otherwise, the multi-component reactions (MCR) of hydrazides, dicarbonyl compounds and ammonium acetate also represent a pathway to synthesize 3,5-disubstituted 1,2,4-triazines.4 Nevertheless, considerable scope for improvement remains in terms of the availability of the starting materials, the tedious nature of the process and the poor functional group tolerance.In the past years, our group reported a method to obtain the imidazole skeleton by employing amidines and ketones/nitroolefins.5 To the best of our knowledge, the units of ketones, acetaldehydes, alkynes and alkenes are useful building blocks providing two carbon atoms to construct five- or six-membered rings.6 Based on our previous works, we shifted our attention to develop six-membered nitrogen heterocyclics using simple operations, the high utilization of atoms and good yields. Herein, we report SeO2- or iodine source-catalyzed [4 + 2] domino annulation reactions for making 1,2,4-triazine derivatives without the addition of transition metal catalysts, and the works systematically study the reactions of ketones/alkynes and amidrazones7 to synthesize 1,2,4-triazine derivatives. These strategies may be an highly efficient alternative to the synthesis of 1,2,4-triazine compounds in pharmaceutical chemistry and other fields.
Results and discussion
Initially, we examined the reaction of acetophenone 1a and benzimidohydrazide 2a as a model reaction, as shown in Table 1. A mixture of 1a (0.2 mmol), 2a (0.2 mmol), CuO (0.24 mmol) and I2 (0.24 mmol) were reacted in DMSO under air at 110 °C for 2 h, and the desired product (3aa) was isolated in 14% yield (Table 1, entry 1). The structure was identified by X-ray crystallography (Fig. 1). Encouraged by this result, we continued to explore other iodine sources, but only inferior results were obtained (Table 1, entries 2–4). Noteworthily, when the reaction was completed, we found that the byproduct of 1,2,4-triazole was formed by TLC, and this side reaction had been documented in previous reports.8 In order to avoid the loss of 2a, we then introduced 2a into the reaction system after the acetophenone reacted for 2 hours in the presence of I2. To our delight, a 60% yield of 3aa was obtained (Table 1, entry 5). Other oxidants, such as MnO2, CuO and SeO2, were also evaluated, and SeO2 showed a better catalytic activity (Table 1, entries 6–8). When the reaction time was prolonged to 4 hours before the addition of 2a, the yield improved to 80% (Table 1, entry 9). In the screening of solvents including DMF, toluene, dioxane and H2O, no better results were obtained (Table 1, entries 10–13).
|
|||
|---|---|---|---|
| Entry | Oxidant | Solvent | Yieldb |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.2 mmol), oxidant (1.2 equiv.), solvent (2 mL), 110 °C, under air.b Isolated yield based on 1a.c Iodine source (0.1 equiv.), TBHP (6 equiv.).d 2a was added 2 hours after the reaction was initiated.e 2a was added 4 hours after the reaction was initiated. | |||
| 1 | CuO/I2 | DMSO | 14 |
| 2 | I2 | DMSO | 27 |
| 3 | I2/TBHPc | DMSO | 24 |
| 4 | TBAI/TBHPc | DMSO | 12 |
| 5 | I2d | DMSO | 60 |
| 6 | MnO2d | DMSO | 0 |
| 7 | CuOd | DMSO | 0 |
| 8 | SeO2d | DMSO | 71 |
| 9 | SeO2e | DMSO | 80 |
| 10 | SeO2e | DMF | Trace |
| 11 | SeO2e | Tolene | 12 |
| 12 | SeO2e | Dioxane | 0 |
| 13 | SeO2e | H2O | 0 |

 | ||
| Fig. 1 The X-ray crystal structure of 3aa. | ||
With the optimized conditions in hand, the scope of this domino annulation reaction was expanded. A variety of different substituted ketones 1 were examined under optimal conditions, and the results are summarized in Table 2. The reaction tolerated well both electron-withdrawing and electron-donating groups on the aromatic ring, and the expected products were obtained in moderate to high yields. Generally, the substrates with electron rich groups provided better yields than the ones with electron deficient groups (Table 2, entries 1–10). Furthermore, the steric effect had no remarkable influence on the yields (Table 2, entries 1–6), and the substrates with sterically bulky groups on the ketones, such as 1-(naphthalen-1-yl)ethanone (1m) and 1-([1,1′-biphenyl]-4-yl)ethanone (1n), and with heterocycles, such as 1-(furan-2-yl)ethanone (1o), were well tolerated, affording good yields in the range 75–78% (Table 2, entries 12–14). Somewhat disappointingly, when aliphatic ketones and substrates with amino groups were investigated in the catalytic system, the desired products were not found (Table 2, entries 11 and 15).
|
|||
|---|---|---|---|
| Entry | R1 | Product | Yieldb |
| a Reaction conditions: 1 (0.2 mmol), 2a (0.2 mmol), SeO2 (0.24 mmol), DMSO (2 mL), 110 °C, 2a was added 4 hours after the reaction was initiated.b Isolated yield based on 1. | |||
| 1 | 4-MeO-C6H4, 1b | 3ab | 80 |
| 2 | 3-MeO-C6H4, 1c | 3ac | 77 |
| 3 | 2-MeO-C6H4, 1d | 3ad | 89 |
| 4 | 4-Me-C6H4, 1e | 3ae | 77 |
| 5 | 3-Me-C6H4, 1f | 3af | 85 |
| 6 | 2-Me-C6H4, 1g | 3ag | 73 |
| 7 | 3,4-MeO-C6H4, 1h | 3ah | 75 |
| 8 | 4-F-C6H4, 1i | 3ai | 76 |
| 9 | 4-Cl-C6H4, 1j | 3aj | 72 |
| 10 | 3,4-Cl-C6H4, 1k | 3ak | 69 |
| 11 | 4-NH2-C6H4, 1l | 3al | 0 |
| 12 | 1-Nap, 1m | 3am | 75 |
| 13 | 4-C6H5-C6H4, 1n | 3an | 76 |
| 14 | 2-Furan, 1o | 3ao | 78 |
| 15 | Propyl, 1p | 3ap | 0 |

To further investigate the reaction scope, various amidrazones (2b–h) were employed to react with acetophenone (1a), as shown in Table 3. Those transformations displayed good functional group tolerance, including methyl, methoxyl, fluoro, chloro and bromo groups, and amidrazones with electron-donating or electron-withdrawing groups had no remarkable difference in yields and gave the corresponding 1,2,4-triazines in good yields (Table 3, entries 1–7). Notably, picolinimidohydrazide (2h) also proceeded smoothly and produced a yield of 58% (Table 3, entry 7).
|
|||
|---|---|---|---|
| Entry | R2 | Product | Yieldb |
| a Reaction conditions: 1a (0.2 mmol), 2 (0.2 mmol), SeO2 (0.24 mmol), DMSO (2 mL), 110 °C, 2 was added 4 hours after the reaction was initiated.b Isolated yield based on 1a. | |||
| 1 | 4-Me-C6H4, 2b | 3ba | 85 |
| 2 | 3-Me-C6H4, 2c | 3bb | 79 |
| 3 | 4-MeO-C6H4, 2d | 3bc | 88 |
| 4 | 4-F-C6H4, 2e | 3bd | 75 |
| 5 | 4-Cl-C6H4, 2f | 3be | 74 |
| 6 | 4-Br-C6H4, 2g | 3bf | 82 |
| 7 | 2-Py, 2h | 3bg | 58 |

Inspired by the above results, we shifted our attention to explore the reaction feasibility of alkynes and amidrazones. The reaction conditions were also investigated (for details, see ESI†), and it was found that the reactions of 4a (0.4 mmol) and 2a (0.2 mmol) in the presence of N-iodosuccinimide (NIS) (0.24 mmol) and p-toluenesulfonic acid (TsOH, 0.02 mmol) in DMSO (2 mL) furnished the desired product in a yield of 62% at 110 °C. Then, the substrate scopes were investigated in detail. Arylalkynes with different functional groups, such as methoxyl, methyl, fluoro, chloro, bromo and phenyl groups, could all provide the corresponding products in good yields ranging from 51 to 82% (Table 4, entries 1–9). Notably, the substrates with electron-donating groups tended to attain better yields, and the steric effect was not critical for those transformations (Table 4, entries 7 and 9).
|
|||
|---|---|---|---|
| Entry | R3 | Product | Yieldb |
| a Reaction conditions: 4 (0.4 mmol), 2a (0.2 mmol), NIS (0.24 mmol), TsOH (0.02 mmol), DMSO (2 mL), 110 °C, 2a was added 4 hours after the reaction was initiated.b Isolated yield based on 2a. | |||
| 1 | 4-MeO-C6H4, 4b | 3ab | 82 |
| 2 | 3-MeO-C6H4, 4c | 3ac | 77 |
| 3 | 4-Me-C6H4, 4e | 3ae | 59 |
| 4 | 3-Me-C6H4, 4f | 3af | 67 |
| 5 | 4-F-C6H4, 4i | 3ai | 66 |
| 6 | 4-Br-C6H4, 4r | 3ar | 57 |
| 7 | 4-Cl-C6H4, 4j | 3aj | 60 |
| 8 | 3-F-C6H4, 4s | 3as | 51 |
| 9 | 2-Cl-C6H4, 4t | 3at | 58 |
| 10 | 4-C6H5-C6H4, 4n | 3an | 79 |

In order to investigate the scope and generality of amidrazones, different amidrazones were employed to react with ethynylbenzene (4a). As expected, the reactions of the amidrazone substrates 2a–h and 4a proceeded smoothly to give the corresponding coupling products 3ba–3bg in moderate to good yields (Table 5, entries 1–7, 65–79%). Regardless of the electronic nature, the functional groups had no remarkable impact on the yields. It was noteworthy that the picolinimidohydrazide (2h) also afforded the desired products in a 64% yield (Table 5, entry 7).
|
|||
|---|---|---|---|
| Entry | R2 | Product | Yieldb |
| a Reaction conditions: 4a (0.4 mmol), 2 (0.2 mmol), NIS (0.24 mmol), TsOH (0.02 mmol), DMSO (2 mL), 110 °C, 2 was added 4 hours after the reaction was initiated.b Isolated yield based on 2. | |||
| 1 | 4-Me-C6H4, 2b | 3ba | 78 |
| 2 | 3-Me-C6H4, 2c | 3bb | 77 |
| 3 | 4-MeO-ph, 2d | 3bc | 71 |
| 4 | 4-F-C6H4, 2e | 3bd | 65 |
| 5 | 4-Cl-C6H4, 2f | 3be | 79 |
| 6 | 4-Br-C6H4, 2g | 3bf | 77 |
| 7 | 2-Py, 2h | 3bg | 64 |

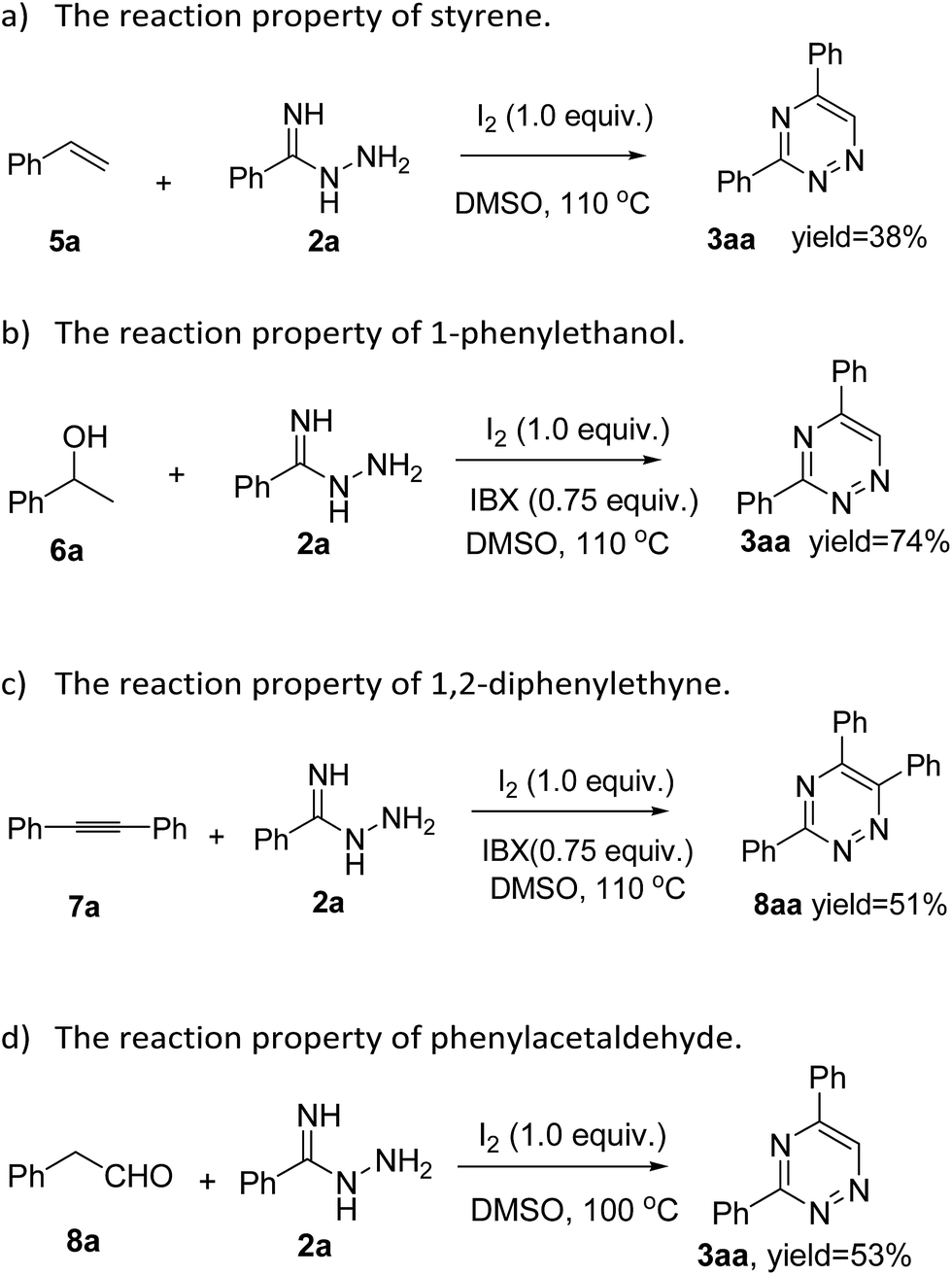
To gain more insight into the diversity of synthetic strategies to produce 1,2,4-triazine derivatives, the building blocks including styrene (5a), 1-phenylethanol (6a), 1,2-diphenylethyne (7a) and 2-phenylacetaldehyde (8a) were introduced into this class of reactions. A reaction of styrene (5a) and 2a was carried out in the presence of iodine (1 equiv.) and DMSO (2 mL) at 110 °C, and the desired product 3aa was isolated in 38% yield (Scheme 1a). Moreover, 1-phenylethanol (6a) and 1,2-diphenylethyne (7a) were reacted with 2a in presence of iodine (1 equiv.), 2-iodoxybenzoic acid (IBX) (0.75 equiv.) and DMSO (2 mL) at 110 °C (Scheme 1b and c), and we were pleased to obtain the corresponding products 3aa and 8aa in 74% and 51% yields, respectively. To our surprise, 2-phenylacetaldehyde (8a) was also compatible in the reaction to form the desired product 3aa in 53% yield (Scheme 1d). These reactions are attractive, making a range of interesting 1,2,4-triazine scaffolds through a useful, efficient and simple procedure by using easily available starting materials.
 | ||
| Scheme 1 Alternative transformations to construct 1,2,4-triazine derivatives starting from styrene, 1-phenylethanol, 1,2-diphenylethyne and phenylacetaldehyde. | ||
Regarding a probable mechanism for these kinds of reactions, some works had reported that the starting materials, such as arylketones9 and arynes,10 converted into corresponding diketones B. Similarly, the arylacetaldehydes, 1-arylethanol11 and alkenes12 might follow a parallel procedure. Subsequently, the diketones combined with amidrazones via a [4 + 2] annulation reaction to obtain the final 1,2,4-triazine products 3 (Scheme 2).13
 | ||
| Scheme 2 Plausible reaction pathway. | ||
Conclusions
In conclusion, we have reported effective and simple strategies to synthesize 1,2,4-triazine derivatives through [4 + 2] domino annulations in one pot. These transformations employ SeO2 or iodine sources as oxidants without using a transition metal catalyst and show good functional group tolerance with moderate to high yields. Moreover, the reactions can access the important 1,2,4-triazine skeleton, which can be potentially applied to afford a series of biologically active derivatives.Experimental
Typical procedure for the preparation of 1,2,4-triazine derivatives 3.Procedure A
The reaction was carried out in a round-bottom side-arm flask (10 mL). 1 (0.2 mmol), SeO2 (0.24 mmol) and DMSO (2 mL) were added to the flask with a magnetic stirring bar at 110 °C under air. After 4 h of stirring at this temperature, 2 (0.2 mmol) was introduced into the catalytic system. 2 hours later, the flask was then removed and cooled to room temperature. The mixture was washed with 20 mL water and extracted with ethyl acetate (3 × 50 mL). The oil layer was combined and concentrated under reduced pressure to distill ethyl acetate, which was further purified by silica gel chromatography (petroleum/ethyl acetate = 5/1 as eluent) to obtain the product 3.Procedure B
The reaction was carried out in a round-bottom side-arm flask (10 mL). 4 (0.4 mmol), NIS (0.24 mmol), TsOH (0.02 mmol) and DMSO (2 mL) were added to the flask with a magnetic stirring bar at 110 °C under air. After 4 h of stirring at this temperature, 2 (0.2 mmol) was introduced into the catalytic system. The following processing method was referred to in procedure A.1H NMR and 13C NMR spectra were recorded on 300 MHz and 75 MHz in CDCl3. Unknown products were further characterized by HRMS (TOF-ESI), and the melting points of solid products were determined on a microscopic apparatus.
Acknowledgements
We are grateful to the project sponsored by the National Science Foundation of P. R. China (No. J11003307 and 21372102).Notes and references
- (a) P. Ahuja and N. Siddiqui, Eur. J. Med. Chem., 2014, 80, 509–522 CrossRef CAS PubMed; (b) M. Anjomshoa, H. Hadadzadeh, M. Torkzadeh-Mahani, S. J. Fatemi, M. Adeli-Sardou, H. A. Rudbari and V. M. Nardo, Eur. J. Med. Chem., 2015, 96, 66–82 CrossRef CAS PubMed; (c) M. Khoshneviszadeh, M. H. Ghahremani, A. Foroumadi, R. Miri, O. Firuzi, A. Madadkar-Sobhani, N. Edraki, M. Parsa and A. Shafiee, Bioorg. Med. Chem., 2013, 21, 6708–6717 CrossRef CAS PubMed; (d) L. Yurttaş, Ş. Demirayak, S. Ilgın and Ö. Atlı, Bioorg. Med. Chem., 2014, 22, 6313–6323 CrossRef PubMed; (e) V. L. Rusinov, I. N. Egorov, O. N. Chupakhin, E. F. Belanov, N. I. Bormotov and O. A. Serova, Pharm. Chem. J., 2012, 45, 655–659 CrossRef CAS; (f) W. P. Heilman, R. D. Heilman, J. A. Scozzie, R. J. Wayner, J. M. Gullo and Z. S. Riyan, J. Med. Chem., 1979, 22, 671–677 CrossRef CAS PubMed; (g) A. Agarwal, K. Srivastava, S. K. Puri and P. M. S. Chauhan, Bioorg. Med. Chem. Lett., 2005, 15, 531–533 CrossRef CAS PubMed.
- (a) F. Marandi, M. Jangholi, M. Hakimi, H. A. Rudbari and G. Bruno, J. Mol. Struct., 2013, 1036, 71–77 CrossRef CAS; (b) E. WoliNska, Tetrahedron, 2013, 69, 7269–7278 CrossRef CAS; (c) F. W. Lewis, L. M. Harwood, M. J. Hudson, A. Geist, V. N. Kozhevnikov, P. Distler and J. John, Chem. Sci., 2015, 6, 4812–4821 RSC; (d) G. L. Guillet, I. F. D. Hyatt, P. C. Hillesheim, K. A. Abboud and M. J. Scott, New J. Chem., 2013, 37, 119–131 RSC; (e) V. Maheshwari, D. Bhattacharyya, F. R. Fronczek, P. A. Marzilli and L. G. Marzilli, Inorg. Chem., 2006, 45, 7182–7190 CrossRef CAS PubMed; (f) M. J. Hudson, C. E. Boucher, D. Braekers, J. F. Desreux, M. G. B. Drew, M. R. S. J. Foreman, L. M. Harwood, C. Hill, C. Madic, F. Marken and T. G. A. Youngs, New J. Chem., 2006, 30, 1171–1183 RSC.
- (a) G. R. Pabst and J. Sauer, Tetrahedron Lett., 1998, 39, 6687–6690 CrossRef CAS; (b) M. Altuna-Urquijo, S. P. Stanforth and B. Tarbitb, Tetrahedron Lett., 2005, 46, 6111–6113 CrossRef CAS; (c) B. M. Culbertson and G. R. Parr, J. Heterocycl. Chem., 1967, 4, 422–424 CrossRef CAS.
- R. Ghorbani-Vaghei, A. Shahriari, Z. Salimia and S. Hajinazari, RSC Adv., 2015, 5, 3665–3669 RSC.
- (a) D. Tang, P. Wu, X. Liu, Y. X. Chen, S. B. Guo, W. L. Chen, J. G. Li and B. H. Chen, J. Org. Chem., 2013, 78, 2746–2750 CrossRef CAS PubMed; (b) X. Liu, D. Wang and B. Chen, Tetrahedron, 2013, 69, 9417–9421 CrossRef CAS; (c) D. Tang, X. L. Li, X. Guo, P. Wu, J. H. Li, K. Wang, H. W. Jing and B. H. Chen, Tetrahedron, 2014, 70, 4038–4042 CrossRef CAS.
- (a) K. K. D. R. Viswanadham, M. P. Reddy, P. Sathyanarayana, O. Ravi, R. Kant and S. R. Bathula, Chem. Commun., 2014, 50, 13517–13520 RSC; (b) Y. Zhu, F. Jia, M. Liu and A. Wu, Org. Lett., 2012, 14, 4414–4417 CrossRef CAS PubMed.
- (a) K. Veejendra, K. Yadavand and G. Babu, Eur. J. Org. Chem., 2005, 2005, 452–456 CrossRef; (b) É. Bokor, A. Fekete, G. Varga, B. Szőcs, K. Czifrák, I. Komáromi and L. Somsák, Tetrahedron, 2013, 69, 10391–10404 CrossRef.
- (a) H. M. Cheng, D. R. Zhu, W. Lu, R. H. Xu and X. Shen, J. Heterocycl. Chem., 2010, 47, 210–213 CrossRef CAS; (b) M. O’rourke, S. A. Lang Jr and E. Cohen, J. Med. Chem., 1977, 20, 723–726 CrossRef.
- (a) H. Z. Li, W. J. Xue and A. X. Wu, Tetrahedron, 2014, 70, 4645–4651 CrossRef CAS; (b) H. Batchu and S. Batra, Eur. J. Org. Chem., 2012, 15, 2935–2944 CrossRef.
- (a) Z. Wan, C. D. Jones, D. Mitchell, J. Y. Pu and T. Y. Zhang, J. Org. Chem., 2006, 71, 826–828 CrossRef CAS PubMed; (b) H. P. Kalmode, K. S. Vadagaonkar and A. C. Chaskar, Synthesis, 2015, 47, 429–438 CrossRef CAS; (c) K. D. R. Viswanadham, M. P. Reddy, P. Sathyanarayana, O. Ravi, R. Kant and S. R. Bathula, Chem. Commun., 2014, 50, 13517–13520 RSC; (d) P. Daw, R. Petakamsetty, A. Sarbajna, S. Laha, R. Ramapanicker and J. K. Bera, J. Am. Chem. Soc., 2014, 136, 13987–13990 CrossRef CAS PubMed; (e) X. F. Xia, Z. Gu, W. Liu, N. Wang, H. Wang, Y. Xia, H. Gao and X. Liu, Org. Biomol. Chem., 2014, 12, 9909–9913 RSC.
- (a) Y. P. Zhu, Q. Cai, Q. H. Gao, F. C. Jia, M. C. Liu, M. Gao and A. X. Wu, Tetrahedron, 2013, 69, 6392–6398 CrossRef CAS; (b) H. P. Kalmode, K. S. Vadagaonkar and A. C. Chaskar, Synthesis, 2015, 47, 429–438 CrossRef CAS.
- S. Dutta, S. S. Kotha and G. Sekar, RSC Adv., 2015, 5, 47265–47269 RSC.
- (a) S. C. Benson, J. L. Gross and J. K. Snyder, J. Org. Chem., 1990, 55, 3257–3269 CrossRef CAS; (b) G. L. Guillet, I. D. Hyatt, P. C. Hillesheim, K. A. Abboud and M. J. Scott, New J. Chem., 2013, 37, 119–131 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1415846. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra26638f |
| This journal is © The Royal Society of Chemistry 2016 |