
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Wool deconstruction using a benign eutectic melt
Katherine E.
Moore
a,
Daniel N.
Mangos
ab,
Ashley D.
Slattery
a,
Colin L.
Raston
*a and
Ramiz A.
Boulos
*a
aCentre for NanoScale Science and Technology, School of Chemical and Physical Science, Flinders University, Bedford Park, South Australia, Australia. E-mail: colin.raston@flinders.edu.au; ramiz.boulos@flinders.edu.au
bThe International Musculoskeletal Research Institute, Adelaide, South Australia, Australia
First published on 9th February 2016
Abstract
Wool fibre is deconstructed in a facile ‘top down’ fabrication process into functional, nano-dimensional α-keratin chains using a benign choline chloride-urea deep eutectic solvent (DES) melt. After breakdown, the keratin can be easily isolated from the DES mixture through dialysis, and freeze-dried to form a protein powder ready for subsequent processing for applications such as wound healing or animal feedstock. The process is simple, efficient, and environmentally friendly. It can potentially utilise what would otherwise be a waste stream, stemming from wool that is deemed unsuitable for the clothing industry, and at the same time providing an additional revenue source.
Introduction
Sheep wool is clearly an abundant biomaterial with applications in the textile industry amongst others. Each year, the wool weaving industry discards tons of non-spin wool fibres that are unsuitable for the textile industry or for making rugs, and this is compounded by the addition of wool-containing human waste such as garments.1 It is therefore desirable, from both economic and environmental standpoints, to eliminate this waste stream and repurpose the waste wool into useful products.Wool consists of three main components: (i) a hydrophobic exterior lipid layer, 18-methyleicosanoic acid (18-MEA) (CH3CH2CH(CH3)-(CH2)15COOH).3 (ii) The outer layer cuticle cells (approximately 0.5 μm thick),4 which point in the direction of the hair tip such that there is no resistance when rubbing a hair fibre in the direction of the follicle to the tip. (iii) The central core, which is composed of a medulla surrounded by a cortex. Fig. 1 shows a schematic diagram depicting the internal structure of a wool fibre and the scale of its components. While the lipid layer is not shown, it is found on the outermost cuticle layer, the cysteine rich epicuticle, and it covalently bound through thioester moieties.5 The single cuticle layer consists of three components, the epicuticle, exocuticle and endocuticle, and differs significantly from human hair, which can comprise up to 7–10 cuticle layers with a total thickness of 4 μm.6 The cortex (visible in the SEM image of Fig. 1) consists of a large number of tightly bound, highly orientated cortical cells, which in turn consist of high sulphur macro- and low sulphur micro-fibrils. The micro-fibrils make up 50–60% by mass of the cortex and are in turn made up of protofibrils, which are composed of the smallest building block of the wool, as highly orientated right-handed α-helix keratin.7 As the wool grows and extends away from the follicle, it undergoes keratinisation where strong intermolecular disulphide bonds form between adjacent keratin strands through oxidation of sulphide groups, –SH. Weaker Coulombic and hydrogen bonding interactions also occur, both intra- and inter-molecularly, giving rise to the strong, rigid and highly ordered wool structure.7 This uniform hierarchical arrangement makes wool an ideal material for ‘top down’ fabrication of the highly useful nanomaterial, keratin. Researchers have achieved this recently using ionic liquids.1,8,9
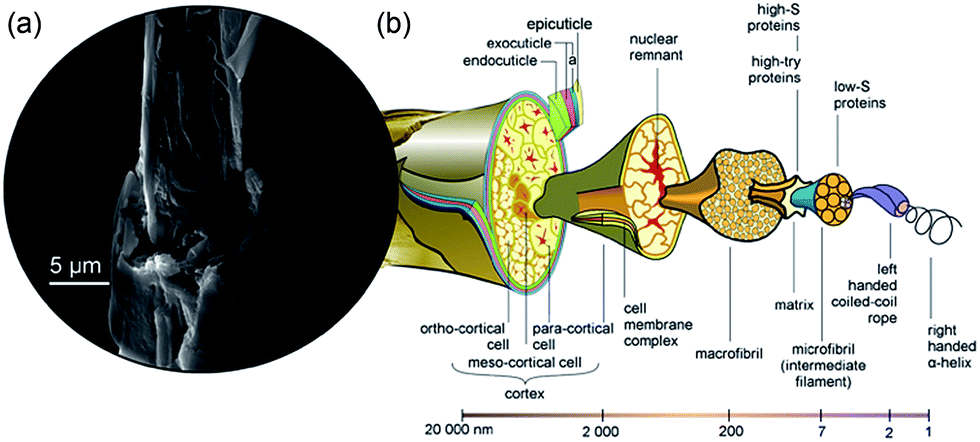 | ||
| Fig. 1 The structure of wool. (a) SEM image of a wool fibre that we stripped of its outer structure revealing a hierarchical structure of cortical cells, macrofibrils and microfibrils. (b) Schematic diagram of a wool fibre showing the internal structure and their corresponding size regimes, adapted with permission from CSIRO.2 | ||
An ionic liquid (IL) is a combination of salts in which the constituent ions are poorly coordinated and unable to form a stable crystal lattice. At least one of the ions has a delocalized charge (in most cases arising from an asymmetrically substituted cation) and one component is organic. As a result, it is a liquid below 100 °C, or even at room temperature, in a way analogous to room temperature ionic liquids (RTILs). To date, there are many useful varieties of IL that can be utilized for natural product extraction, many of which consist of an unsymmetrically substituted cation such as ammonium, imidazolium, pyrrolidinium, phosphonium, and many different constituent anions (especially halogen-based anions) such as Cl−, BrF4−, I−, PF6−, and (CF3SO2)N−.10,11 Interest in ILs arise due to their very low volatility under ambient conditions, solvation prowess for a wide range of inorganic and organic materials and the potential to be highly polar, yet non-coordinating.12 Furthermore, ILs offer good chemical and thermal stability, are non-flammable and highly conductive, and have the ability to stabilize and solubilize a number of organic compounds, including carbohydrates,13–15 feather keratin,16,17 and wood and lignocellulose.18–20 For example, Kilpelainen et al. have demonstrated the successful solubilisation of both hardwoods and soft woods using a number of imidazolium-based ILs over the temperature ranges 80 to 130 °C with dissolving times from 8 to 24 h.19 Most ILs could only partially dissolve wood chips, whereas 1-buty-3-methylimidazolium chloride ([Bmin]Cl) and 1-allyl-3-methylimidazolium chloride ([Amim]Cl) readily dissolve sawdust and thermomechanical pulp fibres. Fully transparent amber solutions can be obtained using 1-benzyl-methylimidazolium chloride ([benzylmim]Cl). This is attributed to the presence of an electron-rich aromatic π-system which can induce stronger interactions with polymers capable of π–π and n–π interactions,19 such as in randomly cross-linked phenylpropanoid units of lignin, which bind the wood network together.21 One major drawback from the use of [benzylmim]Cl however, is its very high viscosity. Highly substituted lignocellulosic esters can be obtained by first dissolving the wood in an IL (130 °C for 4–6 h) and then reacting the resulting mixture with acetyl chloride, benzoyl chloride and acetic anhydride in the presence of pyridine (70 °C for 2 h).20
There have also been several successful applications of IL for the dissolution of wool. For example, Xie et al. employed [Bmim]Cl and [Amim]Cl to dissolve raw wool at 130 °C for 10 h. Similarly, Idris et al. dissolved wool keratin in several types of IL and compared the solubility limit for each. They determine that 1-allyl-3-methylimidazolium dicyanamide ([AMIM][dca]) is the most effective at wool dissolution, solubilising 475 mg of wool per gram of reaction medium at 130 °C over 10 h. Since then, Zheng et al. conducted an analysis of ILs with a number of cation and anion combinations for the dissolution of wool keratin, such as imidazole, pyridine, quaternary ammonium and phosphonium, and acetate, phosphate ester, and halogens, respectively. They determined that for 1-ethyl-3-methylimazolium chloride ([Emim]Cl), the time to dissolve the wool at 130 °C was just 1.5 h.
Strategies have been developed to isolate biomaterials dissolved in ILs, such as in using supercritical CO2 extraction,22 the addition of anti-solvents23 or in chromatography,24 and, simpler options, for example, that presented by Wang and Cao.16 In this work, keratin was extracted from chicken feathers using the eco-friendly and hydrophobic IL, 1-hydroxyethyl-3-methylimidazolium bis(trifluoro-methanesulfonyl)amide ([HOEMIm][NTf2]), with NaHSO3 at 80 °C for 4 h. [HOEMIm][NTf2] posses ‘hyperpolarity’, close to that of protic ILs and water,25 and the NaHSO3 acts to reduce disulphide bonds. After dissolution, water was added and the solution centrifuged to yield a tri-phase system of ionic liquid/parts of insolubilized feathers/keratin. The keratin exhibited good solubility in water while the ionic liquid is immiscible with water, allowing easy separation of the extracted keratin from the reaction system. This process allowed the recovery of the IL with up to 95% efficiency and its reuse for up to five reaction cycles.
Deep eutectic solvents (DESs) are a similar class of reaction media, which share many of the attractive attributes of ILs. They are comprised of a mixture of solid compounds (not necessarily salts), which form a eutectic mixture with a melting point significantly lower than that of either of the individual components.10 Choline chloride and urea is one such DES, which has a melting point of 12 °C (melting points of choline chloride and urea are 303 °C and 133 °C, respectively).26 Charge delocalization is achieved through hydrogen bonding between the halide anion and the amide moiety in urea, and as a result, the compounds experience a decrease in lattice energy, and hence, a decrease in melting point.27 Interestingly, both choline and urea can potentially form hydrogen bonds with the chlorine anion, and may therefore exhibit a synergistic effect.28 Unlike some RTIL, they are easy to prepare in the pure state, are nonreactive with water, biodegradable, benign and affordable.10,26,29,30
To date, the literature has shown many examples where both IL and DES have been used for extraction of a variety of natural products, including phenolic compounds,31 alkaloids,32 essential oils,23,33 organic acids24,34 and carbohydrates14,15 from a range of different biomass materials.
Recently, Boulos et al. demonstrated the use of choline chloride/urea DES as a gentle, benign extraction medium for solubilizing the natural products in human hair.35 In that work, segmented human hair was stirred into a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of choline chloride/urea DES at room temperature overnight, resulting in complete dissolution of the outer lipid layer and isolation of the cuticle cells. They also demonstrated they could repurpose the isolated cuticles and remaining intact cortex as templates for slow-release drug coatings and growth substrates of microalgal cells for wastewater treatment, respectively.
:1 molar ratio of choline chloride/urea DES at room temperature overnight, resulting in complete dissolution of the outer lipid layer and isolation of the cuticle cells. They also demonstrated they could repurpose the isolated cuticles and remaining intact cortex as templates for slow-release drug coatings and growth substrates of microalgal cells for wastewater treatment, respectively.
In the present work, we further exploit the DES extraction outlined by Boulos et al.,35 involving the application of additional heating, directed towards complete deconstruction of low grade waste wool. This not only eliminates a waste product, but yields natural, bioactive keratin suitable for (but not limited to) wound healing and animal feed stock applications.
Materials and methods
The reagents choline chloride and urea were purchased from Sigma-Aldrich. Dialysis membrane tubing with molecular weight cut-off of 6000 kDa was purchased from CelluSep. Raw wool and clean wool that was treated with sodium carbonate and calcined was obtained from Westcoast Wools Pty Ltd (Bibra Lake, Australia).The clean wool was used as obtained, while the raw wool was washed in Milli Q water overnight with stirring to remove any particles from the surface and was then dried at 50 °C for several hours. Fig. 2 shows photographic images of the raw wool before and after washing ((a) and (b) respectively), as well as the clean wool (c). Choline chloride and urea were mixed at a 2:1 molar ratio and stirred with heating (50 °C) to make a eutectic stock solution. To dissolve the wool, 1 g of segmented wool (approximately 1 mm long segments) was added to 20 g of eutectic at 170 °C. The reaction was allowed to proceed for 30 min with stirring to ensure all wool was submerged in the eutectic and allowed to dissolve. The dissolved mixture was able to be stored at 4 °C until needed.
 | ||
| Fig. 2 Photographs of wool prior to DES deconstruction. (a) Raw wool as received and (b) after washing and drying. (c) Commercially treated wool (as received). | ||
For dialysis, equal mass of water was added to the eutectic/keratin mixture post dissolution of the wool, and this allowed dissolution at room temperature, before then being filtered through a membrane with diameter of 11 μm (Whatman Grade 1) to remove any undissolved wool fibres. The filtrate was sealed in the dialysis tubing and kept submerged in frequently changed Milli Q water for 2 days. After dialysis, samples were collected for characterization or frozen at −20 °C prior to freeze-drying at −80 °C at 3 mPa using a Labconco-FreeZone® 1 freeze-drying system.
Chemical analysis
Fourier transform infra-red (FTIR) spectroscopy data was taken with a Nicolet Nexus 870 FT-IR, equipped with a Thermo Scientific ATR-IR ‘Smart Orbit Attachment’. The spectral data was collected at 2 cm−1 resolution with 64 scans at room temperature. Amino acid analysis and liquid chromatography and mass spectrometry were conducted by the Australian Proteome Analysis Facility (APAF) using a Waters AccQtag Ultra Chemistry on a Waters Acquity UPLC (Waters) and a Triple TOF 5600 (AB Sciex) mass spectrometer, respectively.Microscopy techniques
Atomic force microscopy (AFM) measurements were acquired using a Bruker Dimension FastScan AFM with NanoScope V controller, NanoScope control software (version 8.15) and ScanAsyst Air cantilevers (resonant frequency ≈ 70 kHz, spring constant ≈ 0.4 N m−1). The peak-force tapping mode was used with a scan rate of 1 Hz, setpoint of 500 pN, feedback gain of 1.5 and Z-limit of 2.5 μm. Data was analyzed using the NanoScope Analysis software (version 1.4). Scanning electron microscopy (SEM) images were collected with an FEI Inspect F50 Scanning electron microscope operating at 10 keV. Samples were sputter coated with 5 nm of platinum prior to imaging.Results and discussion
In this study, both raw wool, which is surrounded by the 18-MEA lipid layer, and sodium carbonate cleaned wool (referred to as clean wool here after) were investigated. Dissolution of raw wool has the obvious advantage of a simpler processing method with the waste wool directly converted to keratin protein. However, wool contains many environmental contaminants and is therefore likely to require some kind of chemical cleaning procedure prior to processing and subsequent recycling. The sodium carbonate cleaning procedure removes these environmental contaminates as well as the encapsulating lipid layer. Fig. 3(a)–(f) shows SEM images of a raw wool fibre and a clean wool fibre. Qualitatively, the clean fibres look smoother and free of surface contamination; however, more quantitative information about the surface properties can be gained from peak force tapping AFM. Fig. 3(g) and (h) show the surface adhesion forces experienced by a SiO2 tip as it traversed the length of each wool fibre. In each case, ridges corresponding to cuticle edges can be seen, however a uniform surface is seen in the clean sample, which is free of surface contamination. The average adhesion forces observed for the raw and clean samples were approximately 30 and 5 nN, respectively. This significant difference is attributed to the presence of the hydrophobic lipid layer on the raw wool fibre, which increases hydrophobic interactions between the AFM tip and substrate. | ||
| Fig. 3 SEM and AFM images of raw (a–c, g) and clean (d–f, h) wool fibres, respectively. | ||
Raw and clean wool were broken down using the previously reported DES extraction, but with additional heating.35Fig. 4 shows photographs during various stages of processing. Firstly, the wool was dissolved in the DES, forming a viscous amber coloured liquid, as seen in Sample A. The colour arises from melanin pigments nestled between cortical cells, which liberate as the wool fibre dissolves.7
 | ||
| Fig. 4 Photographs at various stages of keratin extraction; namely after dissolution in eutectic (A), after dialysis showing separation of aqueous and precipitated keratin (B), extraction of the aqueous keratin (C) and subsequent freeze drying (D). | ||
Denaturation mechanism
Owing to their growing popularity, there has been an interest in understanding the dissolution mechanisms behind the interaction of ILs and DES, and biological macromolecules. Overall, we can characterize the complex multitude of interactions in ILs and DESs in terms of hydrogen bonding and van der Waals interactions.11 For ILs, the identity of the anion largely determines the extraction efficiency, as solutes are strongly solvated principally through hydrogen bonding between the solute and anion.36 This has been investigated by Kahlen et al. through the use of a Conductor-like Screening Model for Realistic Solvation (COSMO-RS).37 With reference to cellulose, dissolution was possible by disrupting the intra-molecular hydrogen bonding by the solvent. As the interactions of cellulose and IL compete with interactions between ions in the IL, the dissolving power is expected to be strong when one of the ions is highly polar and able to act as a hydrogen donor or acceptor, and the other is only slightly polar. In this respect, only ILs containing anions with a strong hydrogen bond accepting ability are effective, particularly those with a hydrogen bonding basicity, noting a β value of approximately 1, such as for Cl−, are capable of disrupting hydrogen bonds.11 An example is seen in the work of Xie et al.,1 where the substitution of Cl− for Br−, BF4− or PF6− in the IL, 1-butyl-3-methyllimidazolium chloride (BMIM+ Cl−), lead to a reduction in dissolving power towards wool keratin. The same applies to DESs formed between choline chloride and oxalic acid/citric acid in the dissolution of starch.15 The cation identity also effects efficacy, but to a lesser extent. For instance, the length, symmetry and presence of electron rich aromatics affect the viscosity, melting point and capability in forming π–π and π–n bonds, respectively.11While mechanistic studies on ILs and DESs shed light on the breakdown reaction observed here, the case of a choline chloride/urea DES is unusual. Not only is there a highly polar, hydrogen bond accepting anion capable of disrupting intra-molecular bonds, but there is also a high concentration of urea, which is a well establishes denaturant.38–40 Despite its use for more than a hundred years, its mode of function still remains controversial, despite extensive experimental and theoretical studies.40–42 Its denaturing power can be explained by two different proposed mechanisms. The ‘indirect’ mechanism suggests that urea denatures proteins by disrupting the encapsulating hydration layer, which then weakens the hydrophobic interactions, making the hydrophobic residues less compact and more readily solvated.43 The ‘direct’ mechanism suggest that protein unfolding occurs through direct interaction of urea through stronger electrostatic interactions44 with backbone and/or polar residues or through preferential van der Waals interactions42 with the protein. Recently, Zhou et al.38 determined that urea denaturises protein through a two-step kinetic process. This is firstly, by forming a ‘dry molten globule’, which is characterised by a high degree of secondary structure and an extensively disrupted tertiary structure. This leads to a loosely packed conformation with a fluctuating core that unfolds non-cooperatively.45 Secondly, the molten globule is unfolded through preferential binding of urea via van der Waals interactions and water exposure.38
In the case of a choline chloride/urea DES, there is a precedent for a mechanistic study on protein denaturation, focusing on the thermal unfolding and refolding of lysozyme.45 In that work, the authors investigated the protein destabilisation effects of choline chloride/urea and choline chloride/glycerol DESs. They concluded that at high concentration (i.e. pure DES) the protein unfolding was not a two-state process, as observed in MES buffer, where interruption of the secondary and tertiary structure occurs concurrently. Instead, the proteins consisted of partly folded intermediates with a high degree of secondary structure, but with a disrupted tertiary conformation. Unlike the characteristic definition of a molten globule discussed previously for aqueous urea denaturation, the lysozyme intermediates observed by Esquembre et al. had a very compact hydrophobic core.45 The authors also concluded that the choline chloride/urea DES exerted the greatest extraction ability compared to its glycerol counterpart, which is most likely due to the inclusion of a second denaturing agent, urea.
In reference to the literature discussed, the breakdown of wool observed in our work is likely due to disruption of the intra-molecular hydrogen, disulphide and electrostatic bonds by the highly polar DES anion, Cl−, and the urea. Fig. 5 shows SEM images of wool fibres during various stages of breakdown. Fig. 5(a) shows two fibres, one of which is mostly intact with cuticles present on half of the observed fibre. The other has undergone considerably more denaturation with no cuticles and an exposure of cortex cells. Higher magnification images of the left fibre (Fig. 5(b) and (c)) reveal the exposed cortical cells. In Fig. 5(d)–(f), the complete breakdown process of clean wool fibre can be observed on a single fibre. At lower magnification (Fig. 5(d)), cuticles can be seen in the top right of the image, however they are quickly removed and the cortex is revealed. Fig. 5(e) and (f) show that the cortical cells subsequently unravel, revealing the smaller macrofibril structures. These, in turn, unravel into microfibrils and then protofibrils and eventually keratin chains, as the observed amorphous protein material. Fig. 5(g)–(i) further shows an exposed raw wool cortex with a single cuticle remaining after DES melt treatment.
 | ||
| Fig. 5 SEM images of clean (a–f) and raw (g–i) wool, highlighting the various stages of dissolution; namely cuticle removal then exposure and subsequent unravelling of the cortical cells. | ||
Isolation and extraction
While both urea and choline chloride are benign, and being used in cosmetics and as animal feedstock, the high urea concentrations present in the DES might present a safety issue. To address this, the dissolved wool DES mixture was then dialysed against a large volume of Milli Q water for several days in order to reduce the concentration of the DES to appropriate levels. This also resulted in precipitation of keratin that was not completely dissolved and the large melanin pigments, as seen in Sample B of Fig. 4. Careful extraction and subsequent filtration through an 11 μm membrane to remove any remaining wool fibres yield a uniform, aqueous suspension of keratin protein (Sample C). To further isolate the keratin, the sample then underwent freeze-drying to remove the water and to form a solid keratin powder, which proved advantageous for ease of storage and economical transportation. Fig. 4 shows the resultant shard-like keratin powder (Sample D) formed after solvent extraction. Fig. 6 shows SEM images of the isolated keratin, where uniform amorphous keratin sheets and horns can be observed. From hereon, the powder keratin can be easily suspended in a variety of solvents for repurposing, or simply pelletized as an animal feedstock, for example. | ||
| Fig. 6 SEM images of freeze dried dissolved wool, where the keratin forms sheets and horn-like structures upon removal of solvent. | ||
Chemical characterization
To characterize the chemical composition of the deconstructed wool, FTIR, amino acid analysis and liquid chromatography mass spectrometry were employed. From the FTIR spectra shown in Fig. 7(a) and (c), corresponding to raw and clean wool respectively, a broad band at 3300 cm−1 is immediately apparent in both samples, which corresponds to amide stretches and some hydroxyl stretches (commonly referred to as the Amide A band).46 The raw wool fiber exhibits a series of very sharp, intense peaks (indicated with asterisks) between 3000 and 2750 cm−1 that, while being present in other samples, are more intense due to the high surface concentration of carbonaceous lipid. This supports the AFM data seen in Fig. 3, where significantly higher adhesion forces were experienced for the raw wool sample. During the dissolution process, the lipid desorbed from the wool surface and resulted in a reduction of the peaks; however, the Amide A band has a shoulder and exhibits broadening as the desorbed lipid contributes to an increase in the number of hydrogen bonding environments in the sample.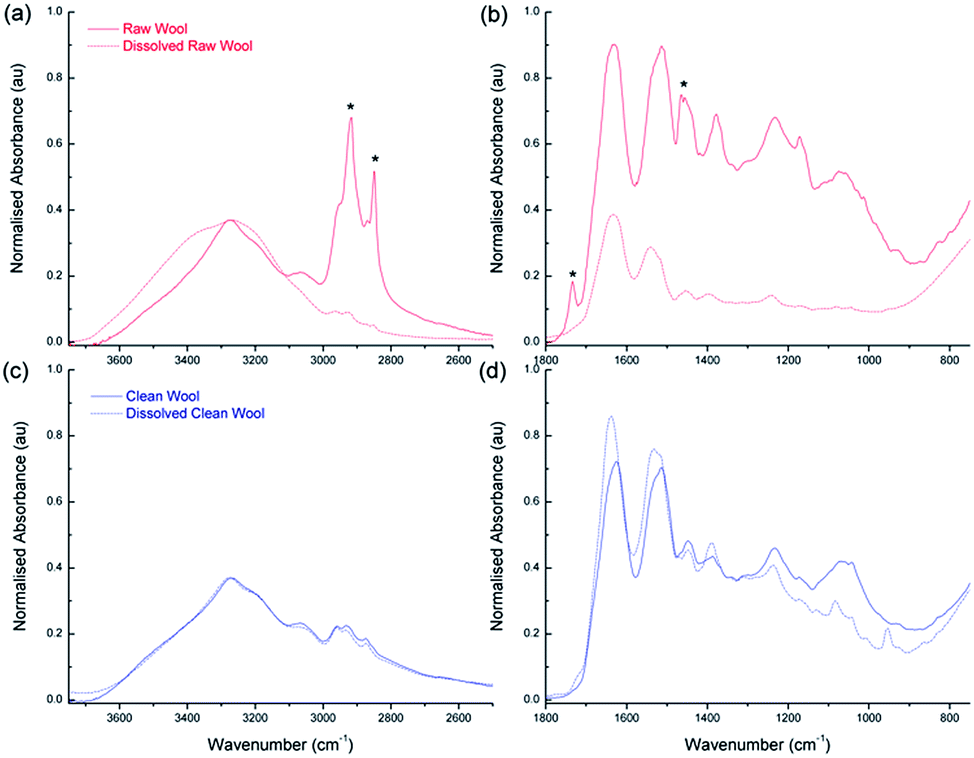 | ||
| Fig. 7 Chemical characterisation of the wool with FTIR spectroscopy. (a) and (b) show high and low wavenumber regions of raw wool before (solid line) and after (dashed line) breakdown using the eutectic melt respectively. The asterisks highlight peaks corresponding to hydrocarbon impurities. Similarly, (c) and (d) shows high and low wavenumber regions for Clean wool before and after treatment using the eutectic melt respectively. FTIR data has been normalized according to an internal standard at 3252 cm−1. | ||
Other large absorption features are seen in Fig. 7(b) and (d) which show the spectra in the region from 2000 to 750 cm−1 for raw and clean wool, respectively. The strongest peak is at 1630 cm−1, commonly referred to as the Amide I peak,46 and corresponds primarily to the stretching vibrations of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (70–85%) and also to C–N groups (10–20%). Amide II is located at 1515 cm−1 and derives mainly from in-plane N–H bending (40–60%). The remaining vibrations arise from the C–N (18–40%) and the C–C (10%) stretching vibrations. Amide III is weak in the FT-IR and is situated at 1235 cm−1. There is also a peak at 1400 cm−1, which corresponds to COO– side chains. This region also shows the presence of hydrocarbons for the raw wool sample with C–H bending and CH2 wagging peaks observed at 1465 and 1170 cm−1, respectively (indicated by asterisks).
O (70–85%) and also to C–N groups (10–20%). Amide II is located at 1515 cm−1 and derives mainly from in-plane N–H bending (40–60%). The remaining vibrations arise from the C–N (18–40%) and the C–C (10%) stretching vibrations. Amide III is weak in the FT-IR and is situated at 1235 cm−1. There is also a peak at 1400 cm−1, which corresponds to COO– side chains. This region also shows the presence of hydrocarbons for the raw wool sample with C–H bending and CH2 wagging peaks observed at 1465 and 1170 cm−1, respectively (indicated by asterisks).
Interestingly, comparison between the raw wool sample and the clean wool shows that the DES breakdown process results in an enhancement of the Amide I peak, with Amide II and Amide III closely maintaining their peak intensity (relative to the internal standard). This indicates that the use of DES affords a purified keratin product, compared to the dissolved raw wool.
Fig. 8 shows the amino acid analysis for dissolved raw and clean samples. The amino acid profile for both wool samples are identical suggesting that the soda ash wash and calcining process had no effect on amino acid content. More importantly, the results show that the process discussed here utilising choline chloride and urea at 170 °C had no effect on the amino acid content. This is a significant finding suggesting the bio-available keratin can be used as a feedstock and/or in cosmetic applications where the amino acid content is necessary for a biological effect, but also for applying relevant quality control criteria to developed products.
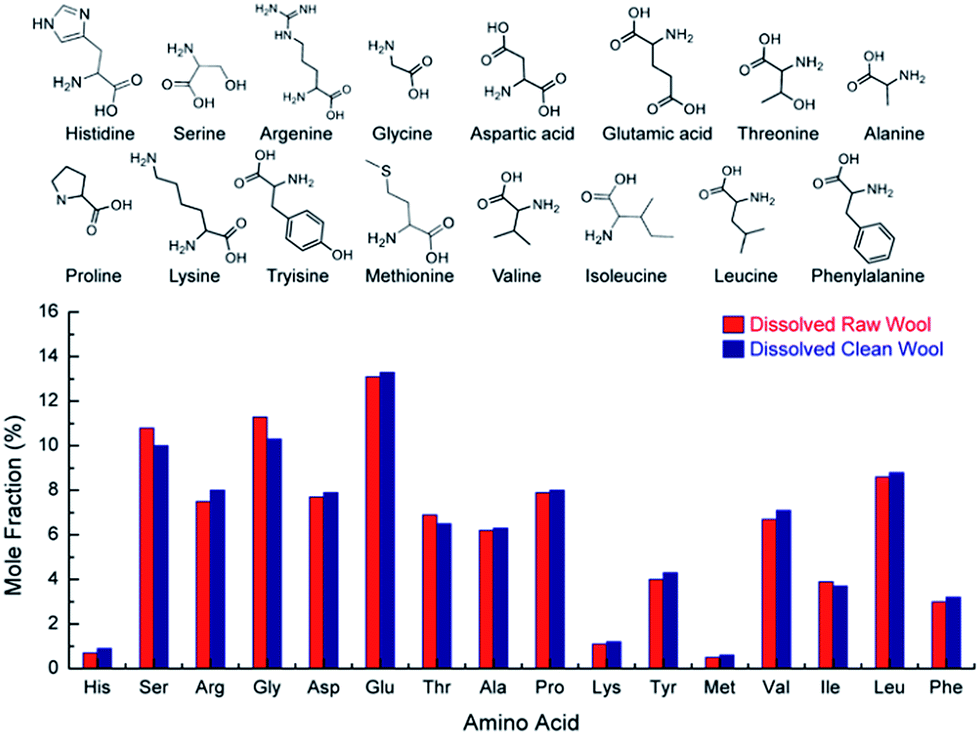 | ||
| Fig. 8 Mole fraction percentage of base amino acids (chemical structures seen above) for dissolved raw and sodium carbonate cleaned wool. | ||
LCMS indicates that the raw and clean extracted keratin consist mainly of Type I microfibrillar sheep keratin with molecular weight of 48 kDa or 47.6 kDa. The samples also include some Type II microfibrillar sheep keratin and small amounts of Type I cuticular sheep keratin.
Conclusions
We have demonstrated a facile process for deconstructing wool for a ‘top down’ fabrication of keratin using a benign eutectic melt. This process can be applied to waste wool that is unsuited for the clothing industry and can therefore eliminate the generation of a waste stream. Importantly, the choline chloride/urea DES breakdown is a green process,47 using only biodegradable and non-toxic chemicals.10,26,29,30 Unlike IL extraction, the DES extracted keratin can easily be isolated from the DES using simple dialysis techniques, and concentrated for subsequent use by freeze drying. The final product would be highly useful for electrospinning to form keratin bandages or for implantation into a hydrogel, for example, both of which have demonstrated clear wound healing advantages.48,49Acknowledgements
The authors wish to acknowledge the Australian Research Council (ARC) and the Government of South Australia for providing funding. The authors also acknowledge the facilities, and the scientific and technical assistance, of the Australian Microscopy & Microanalysis Research Facility (AMMRF) at Flinders Microscopy, Flinders University. Amino acid analysis and LCMS was undertaken at the Australian Proteome Analysis Facility (APAF) with the infrastructure provided by the Australian Government through the National Collaborative Research Infrastructure Strategy (NCRIS).Notes and references
- H. Xie, S. Li and S. Zhang, Green Chem., 2005, 7, 606–608 RSC.
- R. D. Fraser, H. Z. Roe and B. Lipson, The structure of a marino wool fibre, http://www.scienceimage.csiro.au/library/textile/i/7663/the-structure-of-a-merino-wool-fibre/20, 2015 Search PubMed.
- L. N. Jones and D. E. Rivett, Micron, 1997, 28, 469–485 CrossRef CAS PubMed.
- J. Bradbury and J. Leeder, Aust. J. Biol. Sci., 1970, 23, 843–854 CrossRef CAS PubMed.
- J. Swift and J. Smith, J. Microsc., 2001, 204, 203–211 CrossRef CAS PubMed.
- L. J. Wolfram and M. K. Lindemann, J. Soc. Cosmet. Chem., 1971, 22, 839–850 CAS.
- M. Feughelman, Mechanical properties and structure of alpha-keratin fibres: wool, human hair and related fibres, UNSW press, 1997 Search PubMed.
- N. Hameed and Q. Guo, Cellulose, 2010, 17, 803–813 CrossRef CAS.
- R. Li and D. Wang, J. Appl. Polym. Sci., 2013, 127, 2648–2653 CrossRef CAS.
- A. P. Abbott, D. Boothby, G. Capper, D. L. Davies and R. K. Rasheed, J. Am. Chem. Soc., 2004, 126, 9142–9147 CrossRef CAS PubMed.
- Y. Dai, J. van Spronsen, G.-J. Witkamp, R. Verpoorte and Y. H. Choi, J. Nat. Prod., 2013, 76, 2162–2173 CrossRef CAS PubMed.
- T. Welton, Chem. Rev., 1999, 99, 2071–2084 CrossRef CAS PubMed; J. L. Anderson, J. Ding, T. Welton and D. W. Armstrong, J. Am. Chem. Soc., 2002, 124, 14247–14254 CrossRef PubMed.
- L. J. Conceiçao, E. Bogel-Łukasik and R. Bogel-Łukasik, RSC Adv., 2012, 2, 1846–1855 RSC.
- Q. Liu, M. H. Janssen, F. van Rantwijk and R. A. Sheldon, Green Chem., 2005, 7, 39–42 RSC; A. Xu, J. Wang and H. Wang, Green Chem., 2010, 12, 268–275 RSC.
- A. Biswas, R. Shogren, D. Stevenson, J. Willett and P. K. Bhowmik, Carbohydr. Polym., 2006, 66, 546–550 CrossRef CAS.
- Y.-X. Wang and X.-J. Cao, Process Biochem., 2012, 47, 896–899 CrossRef CAS.
- A. Idris, R. Vijayaraghavan, U. A. Rana, D. Fredericks, A. F. Patti and D. R. MacFarlane, Green Chem., 2013, 15, 525–534 RSC.
- U. Domańska and R. Bogel-Łukasik, J. Phys. Chem. B, 2005, 109, 12124–12132 CrossRef PubMed.
- I. Kilpeläinen, H. Xie, A. King, M. Granstrom, S. Heikkinen and D. S. Argyropoulos, J. Agric. Food Chem., 2007, 55, 9142–9148 CrossRef PubMed.
- H. Xie, A. King, I. Kilpelainen, M. Granstrom and D. S. Argyropoulos, Biomacromolecules, 2007, 8, 3740–3748 CrossRef CAS PubMed.
- A. Fujimoto, Y. Matsumoto, H.-M. Chang and G. Meshitsuka, J. Wood Sci., 2005, 51, 89–91 CrossRef CAS.
- M. C. Kroon, J. van Spronsen, C. J. Peters, R. A. Sheldon and G.-J. Witkamp, Green Chem., 2006, 8, 246–249 RSC.
- A. A. Lapkin, P. K. Plucinski and M. Cutler, J. Nat. Prod., 2006, 69, 1653–1664 CrossRef CAS PubMed.
- T. Usuki, N. Yasuda, M. Yoshizawa-Fujita and M. Rikukawa, Chem. Commun., 2011, 47, 10560–10562 RSC.
- S. Zhang, X. Qi, X. Ma, L. Lu and Y. Deng, J. Phys. Chem. B, 2010, 114, 3912–3920 CrossRef CAS PubMed.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70–71 RSC.
- A. P. Abbott, D. Boothby, G. Capper, D. L. Davies and R. K. Rasheed, J. Am. Chem. Soc., 2004, 126, 9142–9147 CrossRef CAS PubMed.
- A. Zhu, T. Jiang, B. Han, J. Zhang, Y. Xie and X. Ma, Green Chem., 2007, 9, 169–172 RSC.
- A. P. Abbott, G. Capper, D. L. Davies, H. L. Munro, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2001, 2010–2011, 10.1039/B106357J.
- D. Coleman and N. Gathergood, Chem. Soc. Rev., 2010, 39, 600–637 RSC.
- F.-Y. Du, X.-H. Xiao and G.-K. Li, J. Chromatogr. A, 2007, 1140, 56–62 CrossRef CAS PubMed; F.-Y. Du, X.-H. Xiao, X.-J. Luo and G.-K. Li, Talanta, 2009, 78, 1177–1184 CrossRef PubMed; L. Zhang and X. Wang, J. Sep. Sci., 2010, 33, 2035–2038 CrossRef PubMed.
- X. Cao, X. Ye, Y. Lu, Y. Yu and W. Mo, Anal. Chim. Acta, 2009, 640, 47–51 CrossRef CAS PubMed.
- Y. Zhai, S. Sun, Z. Wang, J. Cheng, Y. Sun, L. Wang, Y. Zhang, H. Zhang and A. Yu, J. Sep. Sci., 2009, 32, 3544–3549 CrossRef CAS PubMed.
- S. H. Lee, T. V. Doherty, R. J. Linhardt and J. S. Dordick, Biotechnol. Bioeng., 2009, 102, 1368–1376 CrossRef CAS PubMed.
- R. A. Boulos, E. Eroglu, X. Chen, A. Scaffidi, B. R. Edwards, J. Toster and C. L. Raston, Green Chem., 2013, 15, 1268–1273 RSC.
- C. G. Hanke, N. A. Atamas and R. M. Lynden-Bell, Green Chem., 2002, 4, 107–111 RSC.
- J. Kahlen, K. Masuch and K. Leonhard, Green Chem., 2010, 12, 2172–2181 RSC.
- R. Zhou, J. Li, L. Hua, Z. Yang and B. J. Berne, J. Phys. Chem. B, 2011, 115, 1323–1326 CrossRef CAS PubMed.
- W. Li, R. Zhou and Y. Mu, J. Phys. Chem. B, 2012, 116, 1446–1451 CrossRef CAS PubMed; J. F. Brandts and L. Hunt, J. Am. Chem. Soc., 1967, 89, 4826–4838 CrossRef PubMed.
- D. B. Watlaufer, S. K. Malik, L. Stoller and R. L. Coffin, J. Am. Chem. Soc., 1964, 86, 508–514 CrossRef.
- Y. Nozaki and C. Tanford, J. Biol. Chem., 1963, 238, 4074–4081 Search PubMed; A. Caflisch and M. Karplus, Structure, 1999, 7, 477–478 CrossRef CAS PubMed.
- P. Das and R. Zhou, J. Phys. Chem. B, 2010, 114, 5427–5430 CrossRef CAS PubMed.
- H. S. Frank and F. Franks, J. Chem. Phys., 1968, 48, 4746–4757 CrossRef CAS.
- E. P. O'Brien, R. I. Dima, B. Brooks and D. Thirumalai, J. Am. Chem. Soc., 2007, 129, 7346–7353 CrossRef CAS PubMed; M. C. Stumpe and H. Grubmüller, J. Am. Chem. Soc., 2007, 129, 16126–16131 CrossRef PubMed.
- R. Esquembre, J. M. Sanz, J. G. Wall, F. del Monte, C. R. Mateo and M. L. Ferrer, Phys. Chem. Chem. Phys., 2013, 15, 11248–11256 RSC.
- J. M. Cardamone, J. Mol. Struct., 2010, 969, 97–105 CrossRef CAS.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- M. Zoccola, A. Aluigi, C. Vineis, C. Tonin, F. Ferrero and M. G. Piacentino, Biomacromolecules, 2008, 9, 2819–2825 CrossRef CAS PubMed.
- M. Park, H. K. Shin, B.-S. Kim, M. J. Kim, I.-S. Kim, B.-Y. Park and H.-Y. Kim, Mater. Sci. Eng., C, 2015, 55, 88–94 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |