
Chitosan supported silver nanowires as a platform for direct electrochemistry and highly sensitive electrochemical glucose biosensing†
Abstract
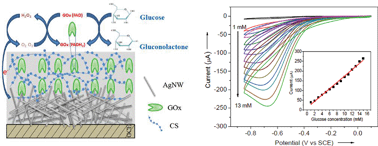
The development of low-cost and sensitive glucose biosensors has been the focus of substantial research interest due to their diverse applications in medical diagnosis, healthcare, and environmental monitoring. Herein, we report the successful use of chitosan (CS) supported silver nanowires (AgNWs) based enzyme electrodes for highly sensitive electrochemical glucose biosensing. The glucose oxidase (GOx) enzyme is electrically contacted using highly conductive AgNWs and thereby significant enhancement in the direct electron transfer (DET) between the redox enzymes and the electrode surface. In addition, the CS polymer matrix enables distinct self-assembly of GOx enzymes adjacent to the electrode surface, which further favours DET by increasing the charge transfer. Characterization by Fourier transform infrared (FTIR) spectroscopy and atomic force microscopy (AFM) measurements were used to evidence the intermolecular interaction and self-assembly of the GOx on CS polymers. AFM results clearly revealed the self-assembly of the GOx on CS surfaces. The immobilized GOx exhibits a well-defined quasi-reversible redox peak with an electron rate constant (ks) of 6.52 s−1 compared to the bare glassy carbon electrode (GCE). The resultant biosensor demonstrates a high sensitivity of 16.72 μA mM−1 cm−2 with a wide linear range (1–15 mM), good selectivity and long-term stability for glucose detection. Our current approach represents a promising platform for the immobilization and electrical wiring of biomolecules with higher loading efficiency for designing low-cost, high sensitive enzymatic biosensors.
 Please wait while we load your content...
Please wait while we load your content...

