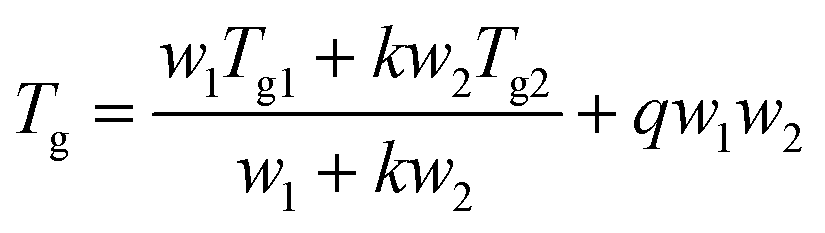DOI:
10.1039/C5RA25982G
(Paper)
RSC Adv., 2016,
6, 8346-8353
Synthesis of a bi-functional terminal polyhedral octasilicate-core dendrimer containing carbazole and 1,8-naphthalimide, and its photoluminescence properties, film formability, and glass transition behavior†
Received
6th December 2015
, Accepted 12th January 2016
First published on 15th January 2016
Abstract
The bi-functional terminal polyhedral octasilicate (OS)-core dendrimer containing carbazole and 1,8-naphthalimide on its peripheries, denoted by OS-NC, was synthesized by the combination of a ring-opening reaction and the condensation reaction of octakis(propenyl succinicanhydro)polyhedral octasilicate (OS-SA) with 9H-carbazole-9-ethanol and N-(hydroxyethyl)-1,8-naphthalimide. The binary blend of the same molar amounts of individual constituent OS-core dendrimers (carbazole-terminated OS-core dendrimer and 1,8-naphthalimide-terminated OS-core dendrimer) was also prepared. The coating film of OS-NC was easily peeled off from the substrate and formed a free-standing film, while the binary blend of the same molar amounts formed a waxy solid. Despite the utilization of intermolecular donor–acceptor interactions, the glass transition temperature (Tg) of the binary blends obeys the Kwei equation with a negative q value. The photoluminescence spectrum of OS-NC and the binary blends in the film state showed exciplex emission. In particular, OS-NC showed a larger red-shift emission than the binary blends.
Introduction
Polyhedral oligomeric silsesquioxanes (POSSs), of the formula R8Si8O12, and a polyhedral octasilicate (OS), of the formula (Si8O20)8−, have attracted much attention in the research field of organic–inorganic hybrids because of their silica-based cubic-like structures with various functional organic substituent groups on their corners.1 POSS- and OS-core dendrimers are particularly attractive because of not only the general advantages of dendrimers, such as well-defined molecular structures and precise molecular weights, but also because of the minimal number of synthetic steps required.2 In addition, the polyhedral structure can inhibit the molecular motion of their branches, with a high proportion of terminal groups positioned on the peripheral surfaces of the dendrimers, even in earlier generations of the dendrimers.3 Thanks to their rigid inorganic cores, the POSS- and OS-core dendrimers have improved mechanical and thermal properties. These aspects indicate that POSS- and OS-core dendrimers terminated with functional units are ideal candidates for the construction of ordered nano-sized platforms for solid materials. Recently, we obtained single component free-standing films based on OS-core dendrimers bearing 16 aromatic groups as the peripheral units.4 Therefore, the POSS- or OS-core dendrimers are suitable for the study of solid state dendrimer materials.
Exciplex formation between donor and acceptor units through the radiative pathway presents a promise in applications such as organic light emitting diodes and sensors.5 The construction of exciplex systems on the surfaces of the OS-core dendrimers would produce novel photoluminescent dendrimer materials with longer emission wavelengths and environmental responsiveness because of effective positioning on their peripheral surfaces. To obtain such solid state dendrimer materials, two approaches are available:6 (1) bi-functional terminal dendrimers which have two kinds of functional groups on the terminal units of the dendrimers,7 and (2) physical blending of two kinds of dendrimers. To the best of our knowledge, however, detailed studies of bi-functional terminal dendrimers and the dendrimer blends for solid state dendrimer materials have not been reported.
Herein, we synthesized a bi-functional terminal OS-core dendrimer by the combination of a ring-opening reaction and condensation reaction of an OS derivative with 8 succinic anhydride groups, using two kinds of simple primary alcohol reagents under mild conditions. As an electron-acceptor, 1,8-naphthalimide shows exciplex emission with electron-donor groups,8 therefore we synthesized the bi-functional terminal OS-core dendrimer bearing 1,8-naphthalimide as an acceptor group and carbazole as a donor group, and also prepared binary dendrimer blends consisting of various ratios of carbazole-terminated and 1,8-naphthalimide-terminated OS-core dendrimers. Moreover, we examined their film forming and thermal properties, and found that the bi-functional terminal OS-core dendrimer formed a free-standing film at room temperature, while the binary dendrimer blend with the same molar amounts of the two kinds of the dendrimers formed no free-standing film. In order to explain the difference in the film formability, the relationship between Tg of the bi-functional terminal dendrimer and the binary dendrimer blends was studied.
Experiment
Materials
All solvents and chemicals were of reagent-grade quality and used without further purification. 9H-Carbazole-9-ethanol (Cz-OH),9 N-(2-hydroxyethyl)-1,8-naphthalimide (Ni-OH),10 octakis(propenylsuccinicanhydrido)polyhedraloctasilicate (OS-SA)11 and carbazole-terminated OS-core dendrimer (OS-Cz)4 were prepared using literature methods.
Measurement
1H (400 MHz), 13C (100 MHz) and 29Si (80 MHz) nuclear magnetic resonance (NMR) spectra were recorded on a Bruker DPX-400 spectrometer (Bruker Biospin GmbH, Rheinstetten, Germany). Ultraviolet-visible (UV-vis) spectra were recorded on a Jasco spectrophotometer V-670 KKN (Jasco, Tokyo, Japan). Fourier transform infrared (FT-IR) spectra were obtained on a JASCO FT/IR-4100 spectrometer (JASCO, Tokyo, Japan) using KBr pellets. Emission spectra were obtained on an FP-8500 (JASCO, Tokyo, Japan), and absolute PL quantum yields (Φ) were determined using a JASCO ILFC-847S. Time-resolved PL lifetime experiments were performed on Quantaurus-Tau (Hamamatsu Photonics). Matrix assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF-MS) was conducted on a Bruker Autoflex II instrument (Bruker Daltonics, Billerica, MA, USA) using α-cyano-4-hydroxycinnamic acid and 2,5-dihydroxybenzoic acid matrices. Differential scanning calorimetry (DSC) was recorded on a TA Instruments 2920 Modulated DSC (TA Instruments, New Castle, DE, USA) under N2. Thermogravimetric analysis (TGA) was performed on a TA Instruments Hi-Res Modulated TGA 2950 thermogravimetric analyzer (TA Instruments) under N2. Gel permeation chromatography (GPC) was measured on a TOSOH LC-8220 (TOSOH, Tokyo, Japan) with TSK-gel α-M column using DMF containing 10 mM LiBr as an eluent, after calibration with standard polystyrenes. Preparative size-exclusion chromatography was employed by a CBM-20A (SHIMAZU, Kyoto, Japan) with KF-2001 and KF-2003 columns, using CHCl3 as an eluent.
9H-Carbazole-carboxylic acid-terminated OS-core dendrimer (OS-Cz-COOH)
OS-SA (1.00 g, 0.468 mmol), Cz-OH (0.810 g, 3.83 mmol), and a catalytic amount of 4-dimethylaminopyridine (DMAP) (50.0 mg, 0.409 mmol) were dissolved in 1,4-dioxane (30 mL). The reaction was allowed to proceed for 12 h at 70 °C, then concentrated and dried under vacuum at 40 °C. The residue was dissolved in CHCl3 and purified by selective precipitation from n-hexane. 1H-NMR (CDCl3): δ 0.04–0.10 (d, 48H, –Si–C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3), 0.43–0.58 (t, 16H, –Si–C2–), 1.19–1.55 (m, 32H, –Si–CH2–C2–C2–), 2.21–2.76 (16H, C2–COO and 8H, C–COO), 4.39–4.47 (m, 16H, O–C2–C2–N), 7.21 (16H, Ar–H), 7.41 (32H, Ar–H), 8.04 (16H, Ar–H). 13C-NMR (CDCl3): δ −0.29, 17.3, 20.4, 35.3, 38.8, 40.7, 41.3, 61.7, 62.1, 108.6, 119.2, 120.3, 122.3, 122.9, 125.8, 126.7, 127.9, 131.1, 131.3, 133.8, 133.9, 140.2, 163.9, 171.6, 174.4. 29Si-NMR (CDCl3): δ −108.9, 12.7.
3), 0.43–0.58 (t, 16H, –Si–C2–), 1.19–1.55 (m, 32H, –Si–CH2–C2–C2–), 2.21–2.76 (16H, C2–COO and 8H, C–COO), 4.39–4.47 (m, 16H, O–C2–C2–N), 7.21 (16H, Ar–H), 7.41 (32H, Ar–H), 8.04 (16H, Ar–H). 13C-NMR (CDCl3): δ −0.29, 17.3, 20.4, 35.3, 38.8, 40.7, 41.3, 61.7, 62.1, 108.6, 119.2, 120.3, 122.3, 122.9, 125.8, 126.7, 127.9, 131.1, 131.3, 133.8, 133.9, 140.2, 163.9, 171.6, 174.4. 29Si-NMR (CDCl3): δ −108.9, 12.7.
Bi-functional terminal OS-core dendrimer containing carbazole and 1,8-naphthalimide (OS-NC)
Purified OS-Cz-COOH was dissolved in dichloromethane (30 mL). To this solution, Ni-OH (1.01 g, 4.21 mmol), DMAP (60.0 mg, 0.491 mmol), and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDAC) (1.61 g, 8.41 mmol) were added. After stirring the mixture at room temperature under N2 for 16 h, the reaction mixture was poured into 1 M HCl. The organic layer was washed with aqueous saturated NaHCO3 brine, dried over MgSO4, and evaporated under vacuum. The residue was dissolved in CHCl3 and purified by selective precipitation from methanol. Further purification was carried out by fractionation with gel permeation chromatography. The yield of pure OS-NC was 1.33 g (51%). 1H-NMR (CDCl3): δ 0.05–0.08 (d, 48H, –Si–C3), 0.40–0.58 (t, 16H, –Si–C2–), 1.09–1.40 (m, 32H, –Si–CH2–C2–C2–), 2.09–2.76 (16H, C2–COO and 8H, C–COO), 4.09–4.34 (m, 16H, O–C2–C2–N), 7.18 (16H, Ar–H), 7.31–7.39 (32H, Ar–H), 7.50–7.59 (16H, Ar–H), 7.88–8.00 (32H, Ar–H), 8.42 (16H, Ar–H). 13C-NMR (CDCl3): δ −0.286, 17.3, 17.4, 20.4, 20.6, 21.7, 35.0, 35.2, 35.3, 35.4, 35.5, 35.6, 38.4, 38.9, 40.7, 40.8, 41.2, 41.4, 61.8, 62.1, 108.6, 119.3, 120.3, 122.3, 122.9, 125.8, 126.8, 128.0, 131.1, 131.4, 133.8, 140.3, 164.0, 171.5, 174.5. 29Si-NMR (CDCl3): δ −108.9, 12.8. MALDI-TOF MS (m/z): calcd for C296H296N16O63Si16Na [M + Na]+: 5638; found: 5638.
1,8-Naphthalimide-termianted OS-core dendrimer (OS-Ni)
OS-Ni was obtained using OS-SA and Ni-OH by following the similar method for OS-NC. The yield of pure OS-Ni was 53%. 1H-NMR (CDCl3): δ 0.04 (d, 48H, –Si–C3), 0.45 (t, 16H, –Si–C2–), 1.21 (m, 16H, –CH2–C2–CH2–), 1.38–1.55 (m, 16H, –CH2–CH2–C2–), 2.33–2.73 (16H, C2–COO and 8H, C–COO), 4.31 (m, 32H, O–C2–C2–N), 7.61 (32H, Ar–H), 8.06 (32H, Ar–H), 8.42 (32H, Ar–H). 13C-NMR (CDCl3): δ −0.34, 17.4, 20.5, 35.2, 35.5, 38.8, 40.7, 61.7, 122.2, 126.8, 127.9, 131.1, 131.3, 133.8, 133.9, 163.8, 163.9, 171.8, 174.5. 29Si-NMR (CDCl3): δ −108.9, 12.9. MALDI-TOF MS (m/z): calcd for C296H280N16O84Si16Na [M + Na]+: 5878; found: 5876.
Preparation of coating film
The solutions of dendrimers were prepared in CHCl3 at 5 wt%. These solutions were casted on a glass or quartz substrate and dried at 3 h at room temperature, and then at 90 °C for 5 min to remove the solvent. The coating films for PL spectra were obtained on a quartz substrate as the same method on the glass substrate.
Results and discussion
Synthesis
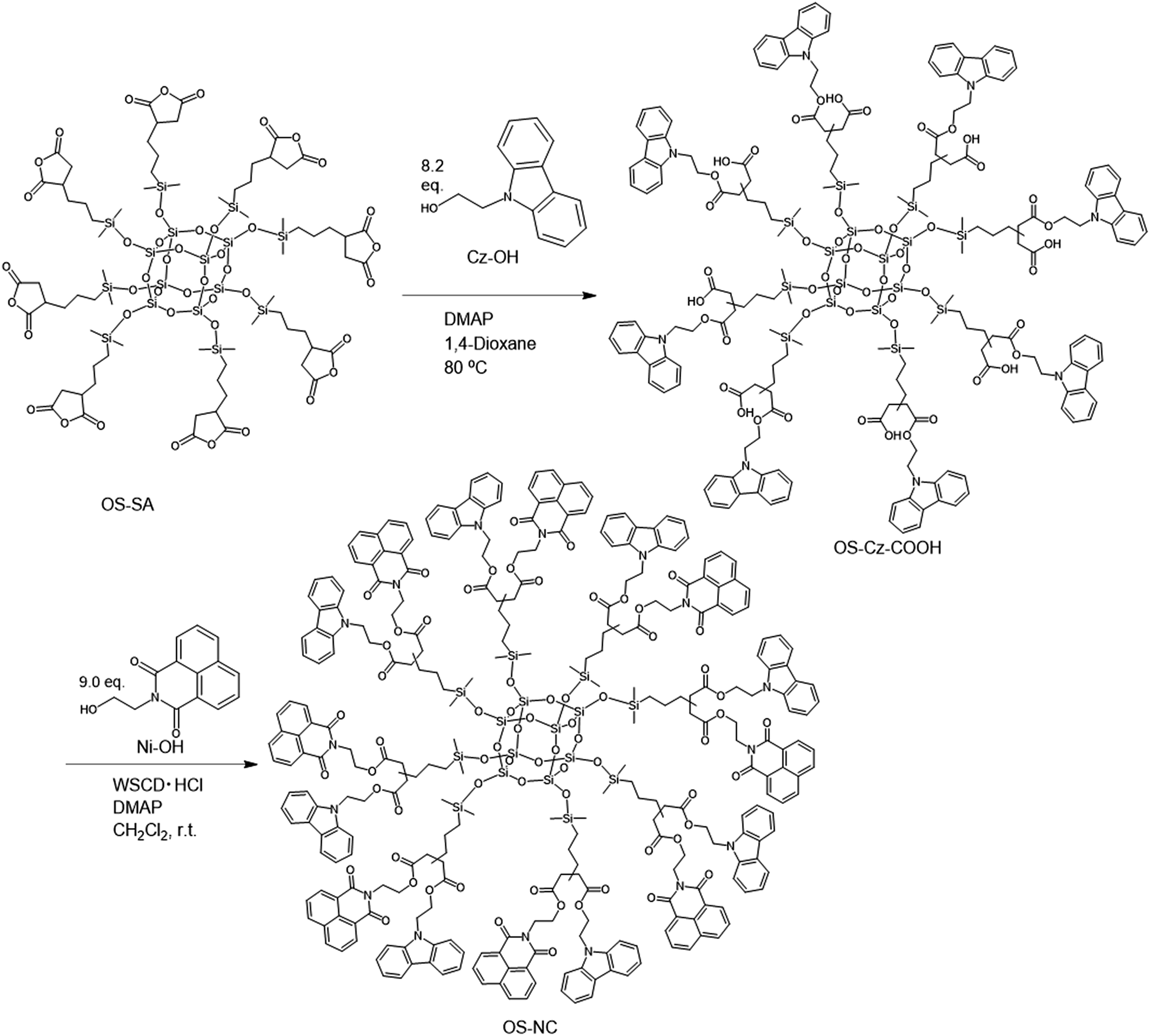
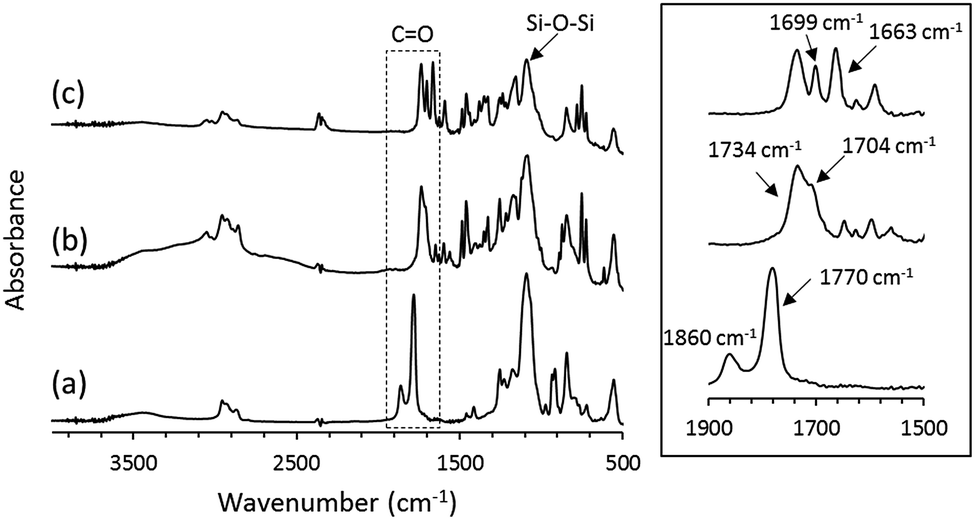
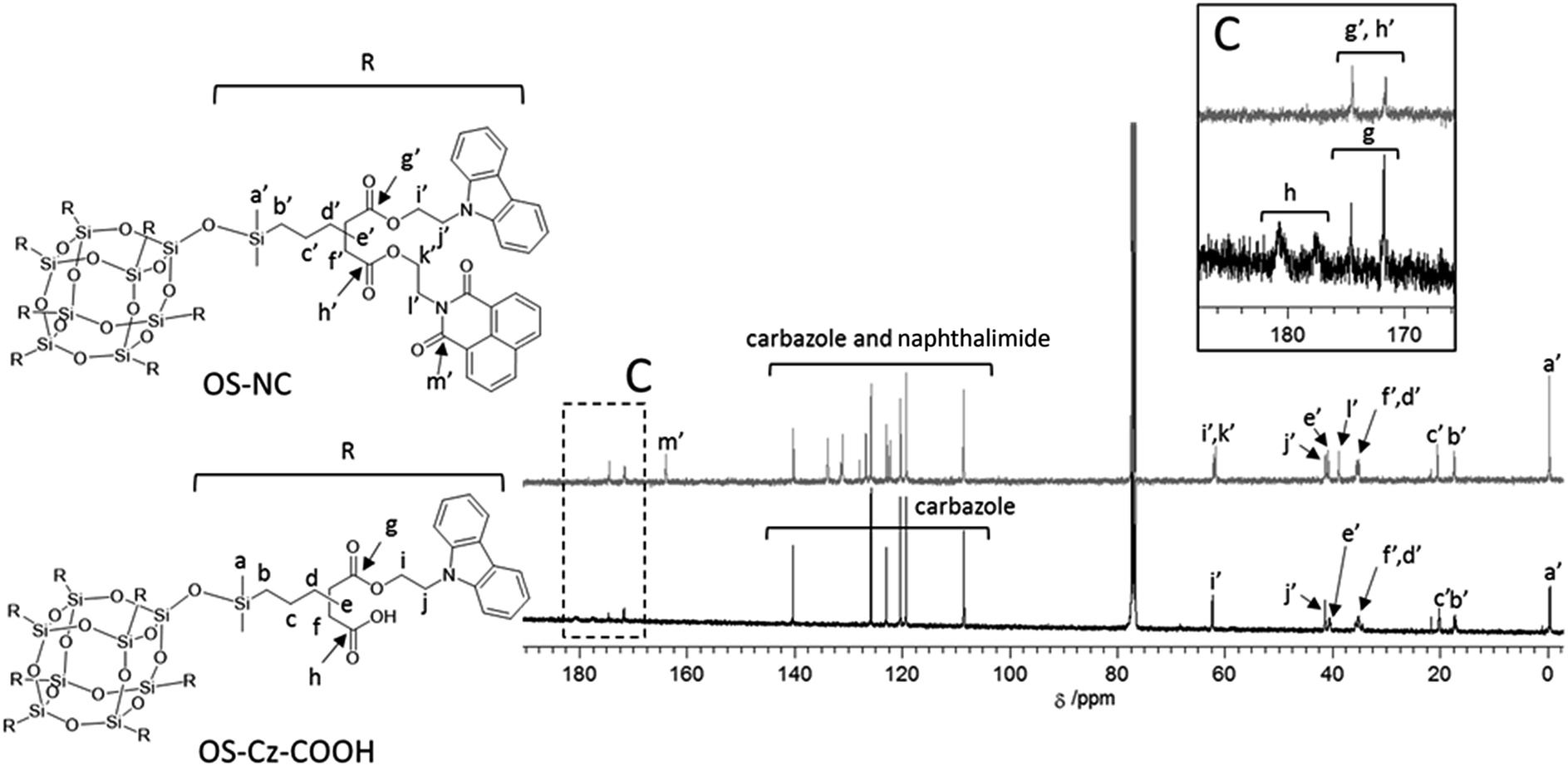
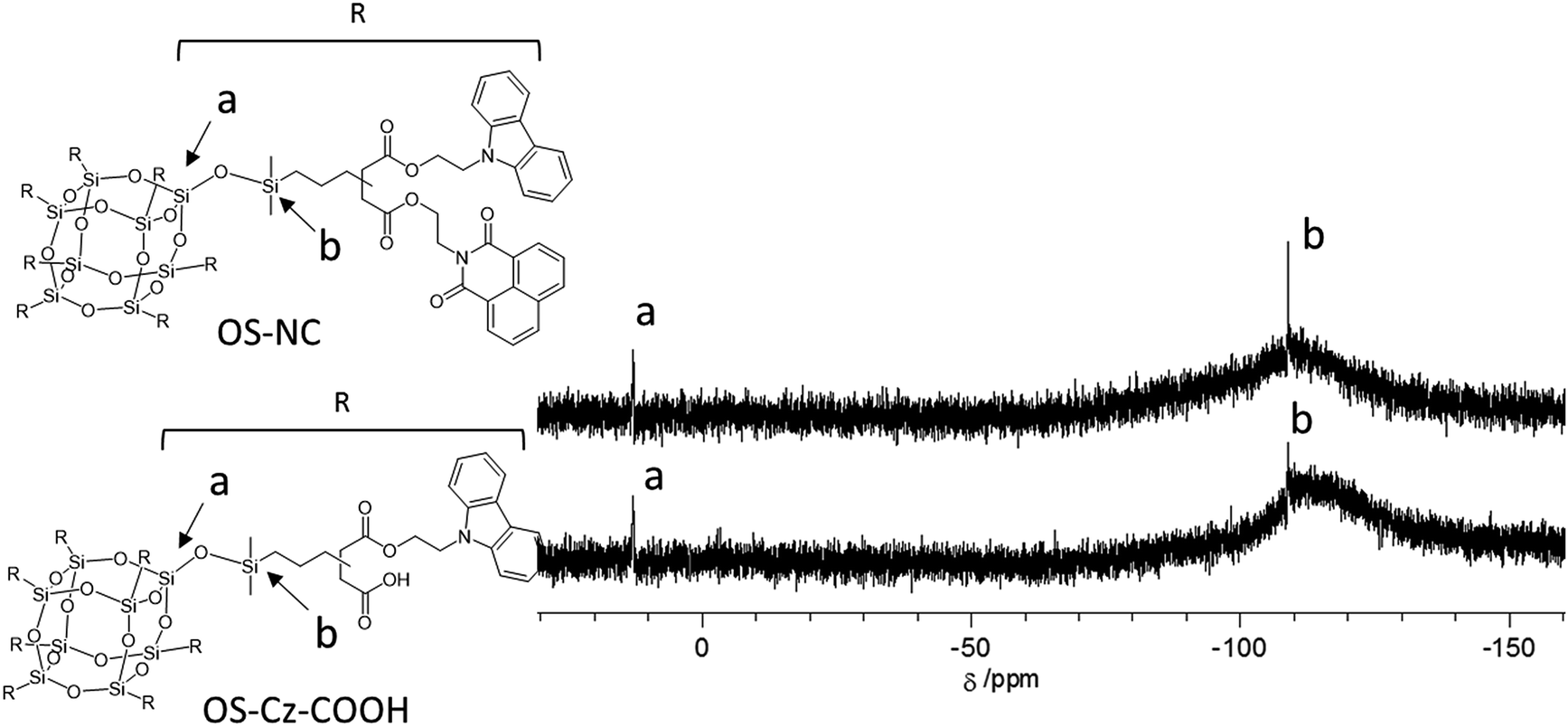
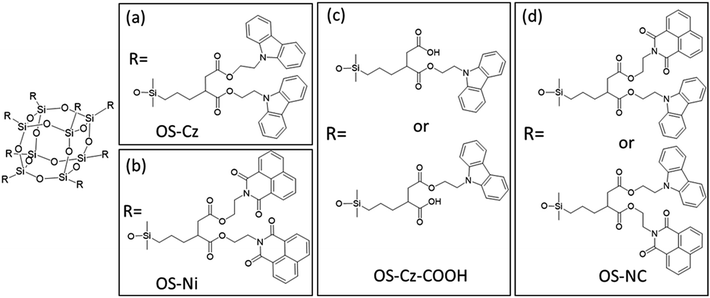
The OS-core dendrimer containing carbazole and 1,8-naphthalimide groups at the terminal group (OS-NC) was synthesized from octakis(propenyl succinicanhydrido)polyhedral octasilicate (OS-SA) by the two step reaction including a ring-opening reaction with 9H-carbazole-9-ethanol (Cz-OH) and subsequent condensation with N-(2-hydroxyethyl)-1,8-naphthalimide (Ni-OH) (Scheme 1). After the first ring-opening reaction, the precursor dendrimer with the carbazole and carboxylic acid groups (OS-Cz-COOH) was purified by multiple sequential precipitations to remove the unreacted compounds. The successful incorporation of the carbazole and carboxylic acid units into OS-Cz-COOH was demonstrated by the characteristic absorption of the carbonyl stretch bands in the FT-IR spectrum (Fig. 1). In the spectrum of OS-Cz-COOH, the stretch band of the acid anhydride in OS-SA at 1860–1770 cm−1 completely disappears and new stretch bands appear, corresponding to the ester and carboxylic acid at 1734 and 1704 cm−1, respectively. The 1H and 13C-NMR spectra further confirmed the structure (Fig. S1 and S2†). The characteristic resonances of the methylene protons adjacent to the OS-core and the aromatic protons on the carbazole groups are observed at δ 0.40–0.54 and 7.21–8.04 ppm, respectively, in the 1H-NMR spectrum. The degree of functionalization of the carbazole groups in OS-Cz-COOH was 48%, estimated by comparison between the areas of the peaks of the aromatic carbazole protons and those of the methylene protons adjacent to the OS core. The 13C-NMR spectrum shows the resonances of the methyl and methylene carbons neighboring the silicon atom, and the aromatic carbazole carbons at −0.283, 17.35, and 108.53–140.35 ppm. In particular, four resonances of the carbonyl carbons at 171.73, 174.59, 177.64, and 180.63 ppm were assigned to two esters and two carboxylic acids in OS-Cz-COOH, indicating the presence of the two kinds of the branching structures that are substituted terminal groups at different positions (Chart 1c). In the 29Si-NMR spectrum of OS-Cz-COOH, two peaks corresponding to the silicate-core and the dimethylsilyl unit for the dendrimer were observed (Fig. 3). No other peaks were detected, indicating no decomposition of the OS core.
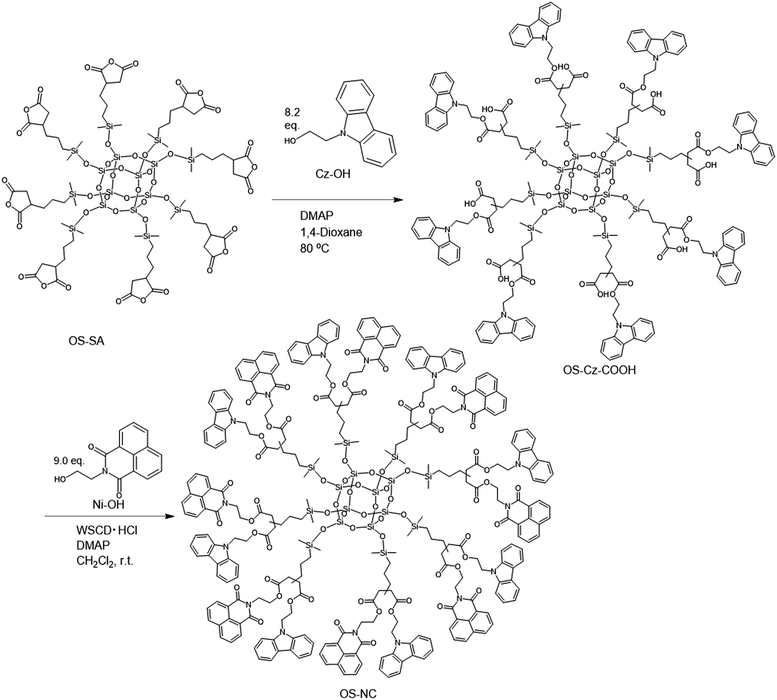 |
| | Scheme 1 Synthesis of OS-NC. | |
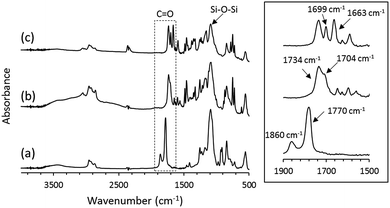 |
| | Fig. 1 (left) FT-IR spectra of (a) OS-SA, (b) OS-Cz-COOH, and (c) OS-NC. (right) Expanded spectra around the carbonyl stretch band. | |
 |
| | Chart 1 Chemical structures of (a) OS-Cz, (b) OS-Ni, (c) OS-Cz-COOH, and (d) OS-NC. | |
OS-Cz-COOH can undergo subsequent functionalization by a condensation reaction with Ni-OH. The structure of the OS-core dendrimer bearing the carbazole and naphthalimide groups (OS-NC) was fully characterized by FT-IR and NMR spectra. In the FT-IR spectrum, the disappearance of the stretch band for the carboxylic acid group and the appearance of two stretch bands for the imide group (1663 and 1699 cm−1) suggest complete conversion from the carboxylic acid in OS-Cz-COOH to the imide group (Fig. 1). In the 1H-NMR spectrum, the peak from the naphthalimide group appears at δ 8.42 and the degree of functionalization of the naphthalimide group was 49%, estimated by comparison between the areas of the peaks of the aromatic naphthalimide protons and those of the methylene protons adjacent the OS-core (Fig. S1†). The degree of total functionalization in OS-NC was 97%. In the 13C-NMR spectrum, the imide carbon of the naphthalimide group can be assigned at 163.95 ppm, and the disappearance of the peaks for the carbonyl carbons of the carboxylic acid at 177.64 and 180.63 ppm (Fig. 2). The spectrum also presented two peaks for the carbonyl carbons of the ester unit, indicating the presence of the bi-functional terminal structure (Chart 1d). To eliminate the probability of the presence of the branching structures having the same terminal groups (i.e. Charts 1a and b) within the dendrimer, the 13C-NMR spectrum of OS-NC was compared with those of OS-Cz and OS-Ni (Fig. S2†). The chemical shifts of the tertiary carbon atoms at the branching point in OS-Cz and OS-Ni appeared in different positions (40.70 and 40.75 ppm, respectively), while that of OS-NC exhibited two signals at different chemical shifts of 40.69 and 40.82 ppm. This observation suggests that OS-NC has only two kinds of tertiary carbons at the branching point. In other words, OS-NC possesses two kinds of terminal group, i.e. carbazole and naphthalimide, respectively, at one branching structure without same terminal group species. Other split peaks such as the methylene proton peak of the adjacent OS core in the 1H-NMR spectrum of OS-NC are caused by these two branching structures. The 29Si-NMR spectrum of OS-NC shows the same two peaks relative to OS-Cz-COOH, indicating no decomposition during the reaction conditions (Fig. 3).
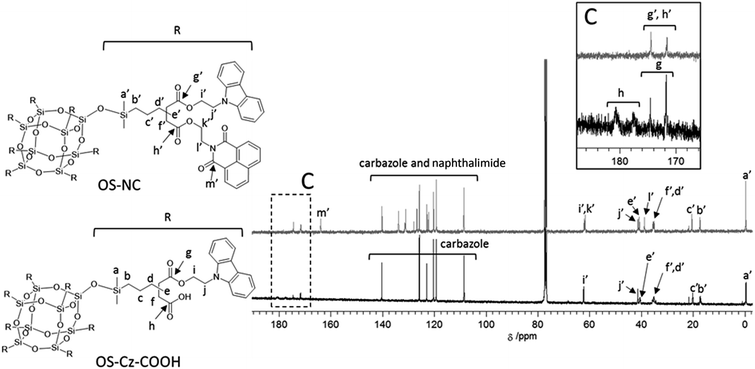 |
| | Fig. 2 13C-NMR spectra, in CDCl3, of OS-COOH (bottom) and OS-NC (top). | |
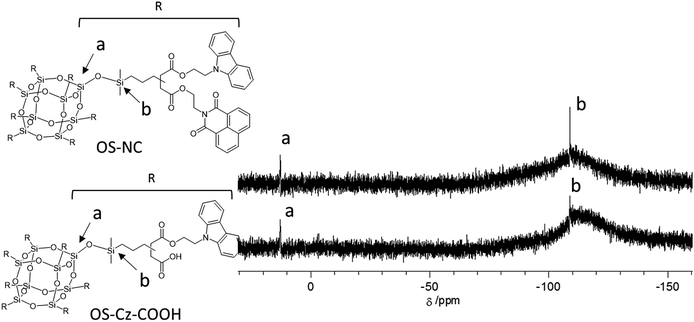 |
| | Fig. 3 29Si-NMR spectra, in CDCl3, of OS-COOH (bottom) and OS-NC (top). | |
The GPC analysis of OS-NC shows a unimodal peak, suggesting purity of the dendrimer (Fig. S3†). The MALDI-TOF mass spectrum of OS-NC showed the presence of peaks at 5638 m/z, which is in agreement with the molecular mass of the bi-functional terminal dendrimer with sodium. Although, the presence of several signals near the molecular mass of OS-NC indicate the bi-functional terminal dendrimer containing different amounts of the carbazole to naphthalimide segments with sodium (Fig. S4†), the GPC analysis and the 13C-NMR spectrum of OS-NC refute the existence of randomly substituted structures. Therefore, we assume that the observation of the several signals occurred due to the MALDI-TOF-MS measurement process.
Optical properties in solution
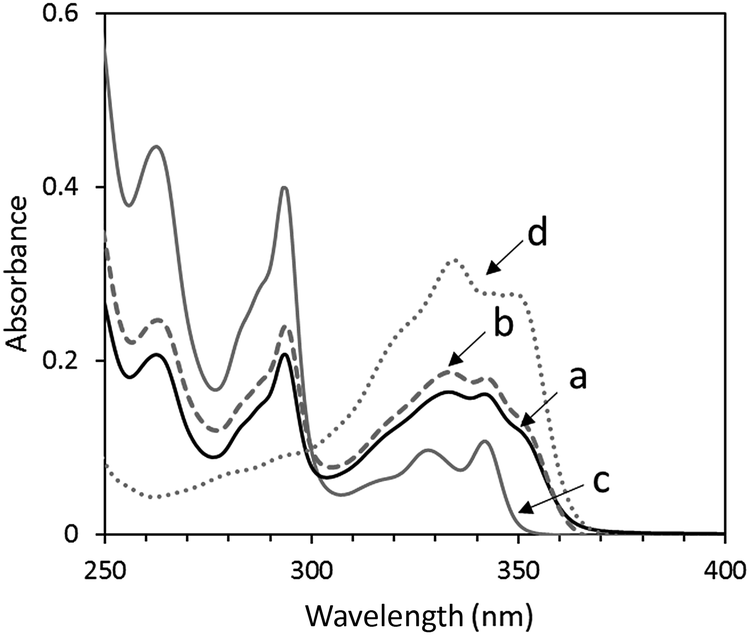
The UV-vis absorption spectrum of the mixture of the same molar amounts (10−6 M) of OS-Cz and OS-Ni in chloroform solution, is the sum of the spectra of OS-Ni and OS-Cz (Fig. 4). Moreover, OS-NC shows the same spectrum as the mixture, indicating no interaction between the carbazole and naphthalimide units in the ground state of OS-NC.
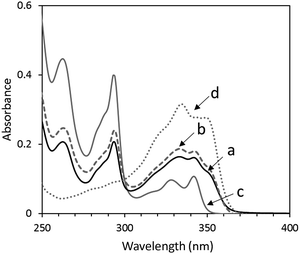 |
| | Fig. 4 UV-vis spectra of (a) OS-NC, (b) the mixture of the same amount of OS-Cz and OS-Ni, (c) OS-Cz, and (d) OS-Ni in chloroform. | |
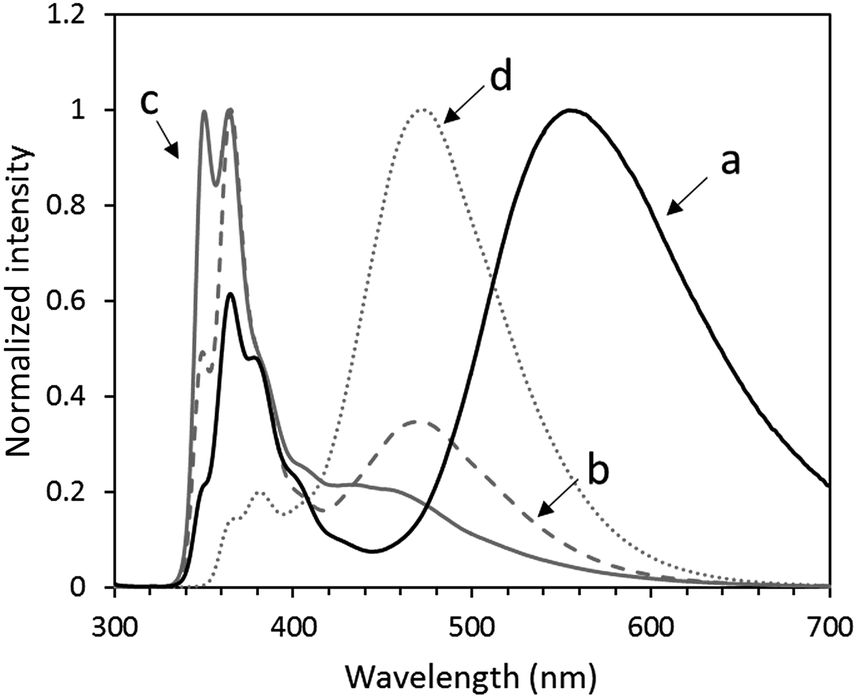
The PL spectrum of the mixture exhibited two PL bands at 364 and 473 nm. The former peak is assigned to the monomer emission of the carbazole and naphthalimide units, and the latter peak is assigned to the excimer emission of the naphthalimide unit (Fig. 5). Furthermore, OS-NC shows a new emission peak at 554 nm in chloroform. However, OS-NC in DMSO shows the monomer emission of the carbazole and naphthalimide units and no new emission peak at 554 nm was detected (Fig. S5†). These results indicate that the emission at 554 nm in the non-polar solvent can be assigned to the PL band due to the exciplex formation between the carbazole and naphthalimide moieties,11 while photoinduced charge separation between the carbazole and naphthalimide units was promoted in the polar solvent. We assume that the carbazole and naphthalimide units in the bi-functional terminal dendrimer are placed at mutual neighbor positions, which leads the exciplex emission in the non-polar solvent. The PL quantum yield of OS-NC in CHCl3 at room temperature was 11.3%.
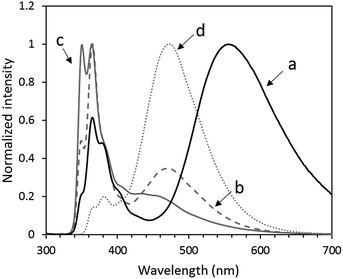 |
| | Fig. 5 PL spectra of (a) OS-NC, (b) the mixture of same amount of OS-Cz and OS-Ni, (c) OS-Cz, and (d) OS-Ni in chloroform, upon excitation at λ = 293 nm. | |
Film formability
The film samples of OS-NC and the mixture of the same molar amounts of OS-Cz and OS-Ni were prepared by casting chloroform solutions of the dendrimers (5 wt%) on glass substrates and subsequent drying at room temperature for 6 h, and then at 90 °C for 5 min to remove the solvent. The thickness of the casting films was 70 μm as estimated by a caliper. We found that both the samples formed yellowish optical transparent films (Fig. 6). The OS-NC film was peeled off from the substrate, resulting in a transparent free-standing film. On the other hand, the film of the binary blend with the same amounts of the individual dendrimers resulted in a waxy solid and no free-standing film was obtained.
 |
| | Fig. 6 Photographs under room light of (A) OS-NC, and (B) free-standing film of OS-NC. | |
Thermal properties
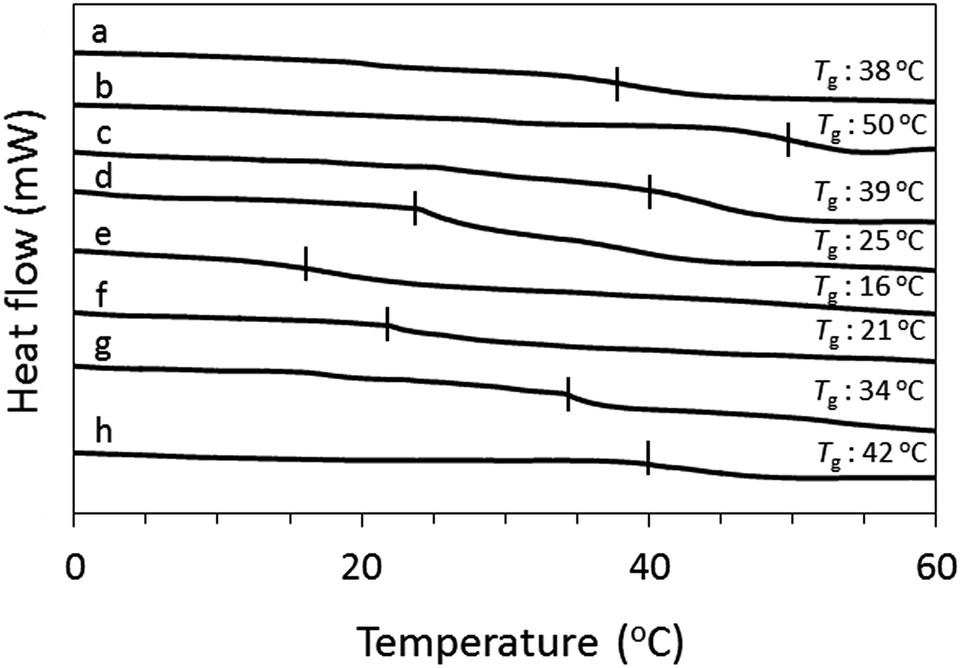
In order to explain the difference in film formability, the thermal behavior of OS-NC, the binary blends of OS-Cz and OS-Ni with various molar ratios, and their individual constituent dendrimers was studied by DSC (Fig. 7). We prepared the binary blends of OS-Cz and OS-Ni with various molar ratios (OS-CxN(1−x); [OS-Ni]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[OS-Cz] = x:[1 − x]). All the binary blends showed only one baseline shift deriving from glass transition (Tg), indicating complete homogeneity. The Tg value of OS-NC was 38 °C, while those of the binary blends decreased, then increased as the molar ratio of OS-Cz increased, after attainment of the minimum temperature.
:[OS-Cz] = x:[1 − x]). All the binary blends showed only one baseline shift deriving from glass transition (Tg), indicating complete homogeneity. The Tg value of OS-NC was 38 °C, while those of the binary blends decreased, then increased as the molar ratio of OS-Cz increased, after attainment of the minimum temperature.
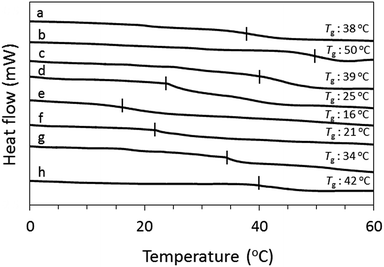 |
| | Fig. 7 DSC curves of (a) OS-NC, (b) OS-Cz, (c) OS-C0.9N0.1, (d) OS-C0.7N0.3, (e) OS-C0.5N0.5, (f) OS-C0.3N0.7, (g) OS-C0.1N0.9, and (h) OS-Ni. | |
The variation of Tg with composition of the binary blends can be predicted by the Kwei equation,12 which considers strong interactions such as hydrogen bonds and charge transfer interactions between different components of binary blends of linear polymers:
where
Tg1, and
Tg2 are the
Tg values of a binary blend of homopolymers 1 and 2, respectively;
w1 and
w2 are the weight fractions of homopolymers 1 and 2;
k is the fitting parameter;
q quantifies the degree of interactions (generally, hydrogen bonding gives a positive
q, and steric hindrance gives a negative
q).
Variations of Tg in the binary blends of OS-Cz and OS-Ni along with fitting from the Kwei equation are shown in Fig. 8. The fitting curve of the Kwei equation shows good agreement with our data, with k = 1 and q = −115. Previous reports describe copolymers and binary blends with charge-transfer interactions which possess positive q values.13 However, our present binary blends show a significantly negative q value. It was reported that physical blends of hyperbranched polymers with terminal hydrogen bonding groups showed a negative q value of −10.6, which was due to the steric hindrance of their terminal groups and the branched structure.6b The unexpectedly large negative q value in our blended systems may be derived from higher degree of steric hindrance due to the highly branched dendrimer structure, and their ample terminal groups, as well as the lack of peripheral movement resulting from the rigid OS core. Thus, the binary blends of the same molar amounts of OS-Cz and OS-Ni caused a large decrease in its Tg value.
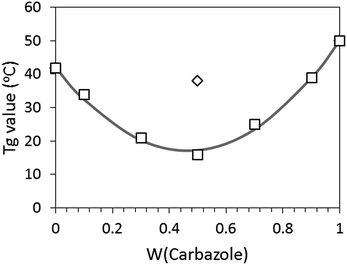 |
| | Fig. 8 (□) Variation of Tg in the binary blends, (◊) Tg of OS-NC, and (solid curve) prediction from the Kwei equation. | |
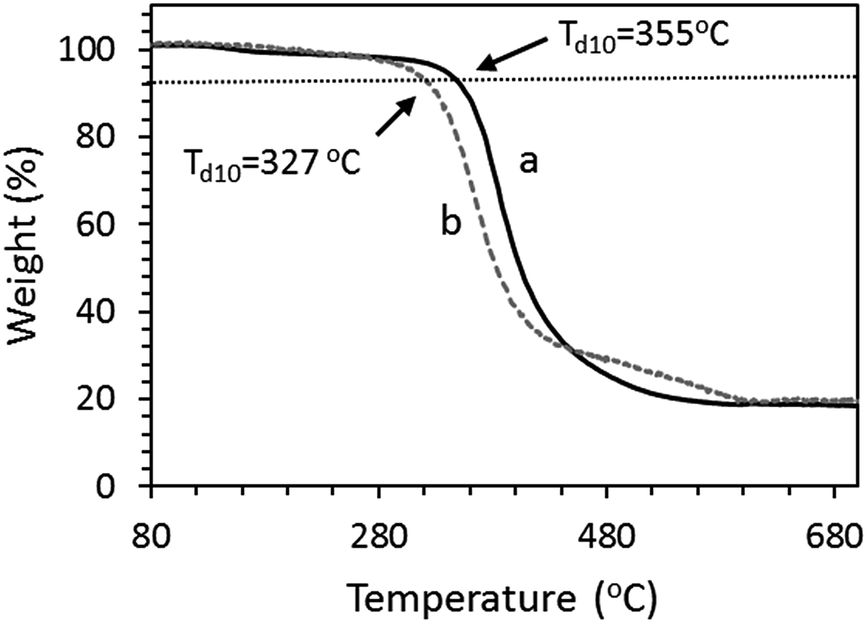
The thermal stabilities of OS-NC and OS-C0.5N0.5 were measured by TGA under N2 atmosphere. OS-NC and OS-C0.5N0.5 showed 10 wt% losses (Td) at 355 °C and 327 °C, respectively (Fig. 9). These results suggest that OS-NC possessed higher thermal stability than OS-C0.5N0.5.
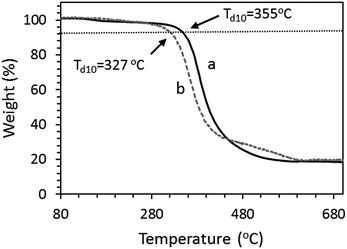 |
| | Fig. 9 TGA thermograms of (a) OS-NC and (b) OS-C0.5N0.5. | |
Optical properties in solid state
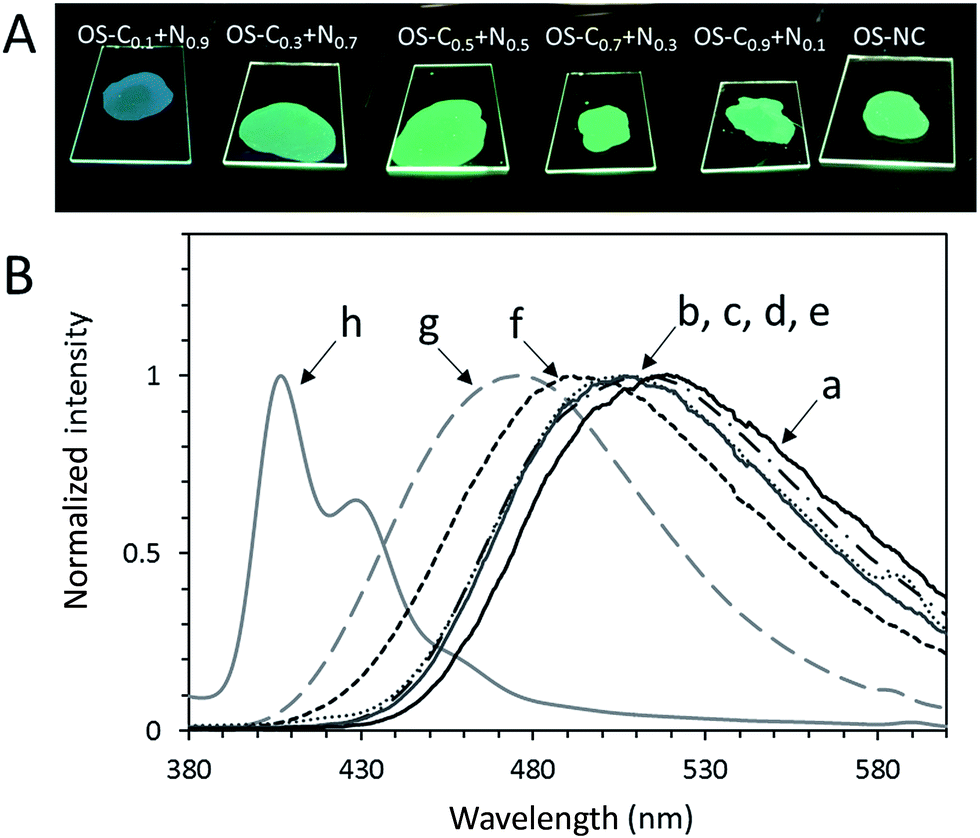
PL spectra and PL quantum yields of OS-NC, OS-Cz, OS-Ni, and the binary blends are shown in Fig. 10 and their maximum emission wavelengths are listed in Table 1. The emission spectra of OS-NC and the binary blends show only one peak, which can be assigned as exciplex emission because it is distinct from those of either OS-Cz or OS-Ni.14 To obtain additional evidence of exciplex formation, we execute time-resolved PL decay measurements in the solid state at room temperature (Fig. S6†). The average lifetimes (τavg) of OS-Cz and OS-Ni are 10 and 26 ns, respectively. On the other hand, OS-NC and OS-C0.5N0.5 showed much longer τavg of 44 and 47 ns, respectively. These results clearly suggests the emissions of OS-NC and the binary blends are originated to the long-lived exciplex.15 The PL spectrum of OS-NC showed a longer maximum emission wavelength (522 nm) than those of the binary blends. The closely substituted carbazole and naphthalimide moieties at the spherical surface in the single component scaffold of OS-NC formed a higher overlapping exciplex compared to the binary blends,16 leading to the largest red-shift emission in OS-NC. On the other hand, the maximum emission wavelengths of the binary blends were observed at 492 nm and 511 nm. Poor reproducibility of the binary blends for the maximum emission wavelength in comparison with that of OS-NC suggests that the molecular-level structure of binary blends was poorly controlled.
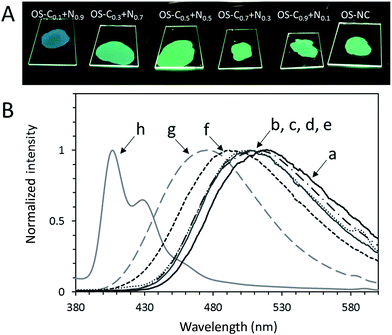 |
| | Fig. 10 (A) Photographs of OS-NC and the binary blends under UV, and (B) PL spectra of (a) OS-NC, (b) OS-C0.9N0.1, (c) OS-C0.7N0.3, (d) OS-C0.5N0.5, (e) OS-C0.3N0.7, (f) OS-C0.1N0.9, (g) OS-Ni, and (h) OS-Cz in the solid state (λex = 293 nm). | |
Table 1 Optical properties of OS-NC and the binary blends
| Sample |
λmaxa [nm] |
QYb [%] |
| Maximum emission wavelength. Absolute quantum yield in solid state. |
| OS-NC |
523.0 ± 0.6 |
7.6 |
| OS-C0.1N0.9 |
495.0 ± 2.2 |
10.0 |
| OS-C0.3N0.7 |
511.0 ± 2.8 |
8.0 |
| OS-C0.5N0.5 |
511.0 ± 2.5 |
8.2 |
| OS-C0.7N0.3 |
511.8 ± 2.5 |
8.9 |
| OS-C0.9N0.1 |
505.2 ± 2.2 |
8.4 |
Conclusions
A novel bi-functional terminal organic–inorganic dendrimer with carbazole and naphthalimide units was successfully prepared by a hydrosilylation reaction, and a combination of a ring-opening reaction and condensation reaction of OS-SA. The coating film of OS-NC can be peeled off from the substrate to give a free-standing film. In the comparison of OS-NC with the various binary blends, based on the results of the DSC measurement, the Tg value of the binary blends was decreased relative to their individual constituent dendrimers. However, the Tg value of OS-NC was shown to be above room temperature. The PL spectra of OS-NC and binary blends in the film state showed exciplex emission. In particular, OS-NC showed a larger red-shift emission than the binary blends.
Acknowledgements
This study is a part of a Grant-in-Aid for Science Research (No. 24350058) from the Ministry of Education, Culture, Sports, Science, and Technology, Government of Japan, and a Grant-in-Aid for Scientific Research on Innovative Areas “New Polymeric Materials Based on Element-Blocks (No. 2401)” (24102003) of The Ministry of Education, Culture, Sports, Science, and Technology, Japan. We thank Prof. Tsuyoshi Kawai of Nara Institute of Science and Technology for measuring MALDI-TOF-MS supported by Nanotechnology Platform. We are also indebted to Prof. M. Shimizu and Mr R. Shigitani of Kyoto Institute of Technology for PL lifetime measurements.
Notes and references
-
(a) Y. Chujo and K. Tanaka, Bull. Chem. Soc. Jpn., 2015, 88, 633–643 CrossRef CAS;
(b) D. B. Cordes, P. D. Lickiss and F. Rataboul, Chem. Rev., 2010, 110, 2081–2173 CrossRef CAS PubMed.
-
(a) J. Yang, Q. Zhang, H. Chang and Y. Cheng, Chem. Rev., 2015, 115, 5274–5300 CrossRef CAS PubMed;
(b) A. Sellinger and R. M. Laine, Chem. Mater., 1996, 8, 1592–1593 CrossRef CAS;
(c) K. Naka, M. Fujita, K. Tanaka and Y. Chujo, Langmuir, 2007, 23, 9057–9063 CrossRef CAS PubMed.
- K. Naka, R. Shinke, M. Yamada, F. D. Belkada, Y. Aijo, Y. Irie, S. R. Shankar, K. S. Smaran, N. Matsumi, S. Tomita and S. Sakurai, Polym. J., 2014, 46, 42–51 CrossRef CAS.
-
(a) Y. Irie and K. Naka, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1437–1443 CrossRef CAS;
(b) Y. Irie and K. Naka, J. Polym. Sci., Part A: Polym. Chem., 2016 DOI:10.1002/pola.27811.
-
(a) T. S. Reddy and A. R. Reddy, J. Fluoresc., 2014, 24, 1571–1580 CrossRef PubMed;
(b) Y. Seino, H. Sasabe, Y.-J. Pu and J. Kido, Adv. Mater., 2014, 26, 1612–1616 CrossRef CAS PubMed;
(c) R.-H. Lee, K.-T. Lin and C.-Y. Huang, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 330–341 CrossRef CAS;
(d) T. Offermans, P. A. van Hal and S. C. J. Meskers, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 045213 CrossRef.
-
(a) W. Brostow, R. Chiu, I. M. Kalogeras and A. Vassilikou-Dova, Mater. Lett., 2008, 62, 3152–3155 CrossRef CAS;
(b) T. Liu, X. Geng, Y. Nie, R. Chen, Y. Meng and X. Li, RSC Adv., 2014, 4, 30250–30258 RSC.
-
(a) P. Antoni, Y. Hed, A. Nordberg, D. Nyström, H. V. Holst, A. Hult and M. Malkoch, Angew. Chem., Int. Ed., 2009, 48, 2126–2130 CrossRef CAS PubMed;
(b) A. P. Goodwin, S. S. Lam and J. M. J. Fréchet, J. Am. Chem. Soc., 2007, 129, 6994–6995 CrossRef CAS PubMed;
(c) X.-X. Deng, F.-S. Du and Z.-C. Li, ACS Macro Lett., 2014, 3, 667–670 CrossRef CAS.
-
(a) T. Miyazaki, M. Shibahara, J. Fujishige, M. Watanabe, K. Goto and T. Shinmyozu, J. Org. Chem., 2014, 79, 11440–11453 CrossRef CAS PubMed;
(b) D. W. Cho, M. Fujitsuka, K. H. Choi, M. J. Park, U. C. Yoon and T. Majima, J. Phys. Chem. B, 2006, 110, 4576–4582 CrossRef CAS PubMed.
- H. Duana, L. Liua, X. Menga, D. Qina, X. Wanga, X. Dua, C. Wangb and J. Tianc, Synth. Met., 2009, 159, 1512–1516 CrossRef.
- J. Malinge, C. Allain, A. Brosseau and P. Audebert, Angew. Chem., Int. Ed., 2012, 51, 8534–8537 CrossRef CAS PubMed.
- S. Skaria and S. Schricker, J. Macromol. Sci., Part A: Pure Appl.Chem., 2010, 47, 381–391 CrossRef CAS.
- T. K. Kwei, J. Polym. Sci., Polym. Lett. Ed., 1984, 22, 307 CrossRef CAS.
-
(a) A. Simmons and A. Natansohn, Macromolecules, 1990, 23, 5127–5132 CrossRef CAS;
(b) J. M. Rodriguez-Parada and V. Percec, Macromolecules, 1986, 19, 55–64 CrossRef CAS.
-
(a) L. C. Palilis, A. J. Mäkinen, M. Uchida and Z. H. Kafafi, Appl. Phys. Lett., 2003, 82, 2209–2211 CrossRef CAS;
(b) A. C. Morteani, P. Sreearunothai, L. M. Herz, R. H. Friend and C. Silva, Phys. Rev. Lett., 2004, 92, 247402 CrossRef PubMed.
-
(a) J. J. Benson-Smith, J. Wilson, C. Dyer-Smith, K. Mouri, S. Yamaguchi, H. Murata and J. Nelson, J. Phys. Chem. B, 2009, 113, 7794–7799 CrossRef CAS PubMed;
(b) C. Dyer-Smith, J. J. Benson-Smith, D. D. C. Bradley, H. Murata, W. J. Mitchell, S. E. Shaheen, S. A. Haque and J. Nelson, J. Phys. Chem. C, 2009, 113, 14533–14539 CrossRef CAS;
(c) J. H. Kim, B.-K. An, S.-J. Yoon, S. K. Park, J. E. Kwon, C.-K. Lim and S. Y. Park, Adv. Funct. Mater., 2014, 24, 2746–2753 CrossRef CAS.
- K. Nagura, S. Saito, H. Yusa, H. Yamawaki, H. Fujihisa, H. Sato, Y. Shimoikeda and S. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 10322–10325 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and spectrum data. See DOI: 10.1039/c5ra25982g |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.