
In vivo mapping and assay of matrix metalloproteases for liver tumor diagnosis†
Yu-Feng Chen‡
a,
Jin Hong‡b,
Dong-Yan Wua,
Ying-Ying Zhoua,
Mathew D'Ortenzioc,
Ya Ding*a and
Xing-Hua Xiad
aKey Laboratory of Drug Quality Control and Pharmacovigilance, China Pharmaceutical University, Ministry of Education, Nanjing 210009, China. E-mail: ayanju@163.com
bKey Laboratory of Biomedical Functional Materials, School of Sciences, China Pharmaceutical University, 639 Longmian Avenue, Nanjing 211198, China
cUniversity of Waterloo, 200 University Avenue West, Waterloo, ON N2L 3G1, Canada
dKey Lab of Analytical Chemistry for Life Science, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, China
First published on 14th January 2016
Abstract
To improve current cancer treatment efficacy, a method for early and accurate detection is of great importance, especially for cancer biomarkers with a specific theranostic target. Matrix metalloproteases (MMPs) are a family of endopeptidases that are highly expressed in various types of cancers and play an important role inmetastasis. To detect and locate over-expressed MMPs in vitro and in vivo, a fluorescent “off–on” nanoswitch (GNP-p-FITC/mPEG) has been developed by engineering the fluorescein isothiocyanate-labeled peptide (p-FITC) and thiolated polyethyleneglycol monomethyl ether (HS-mPEG, molecular weight 2000 Da) on the surface of gold nanoparticles (GNPs). The p-FITC contains a MMP preferential cleavage site and HS-mPEG is used to improve the stability and biocompatibility of the nanoswitch in vivo. The nanoswitch has a very low background signal and displays sensitive response to compound enzymes (containing MMP-2, MMP-4, and MMP-9 that may contribute to MMPs activity) either in vitro or in vivo. As a result of using GNPs, the nanoswitch has enhanced cellular internalization with HepG2 cells due to the endocytosis process as well as improved tumor penetration performance due to its small-size-induced enhanced permeability and retention (EPR). Furthermore, the biocompatibility and tumor targeting ability of the nanoswitch are established by its cytotoxicity with HepG2 cells and distributions in normal organs and tumor tissues. The simple preparation process, the complete fluorescence quenching, and the effective fluorescence recovery (about 50-fold intensity) makes the present MMP-responsive nanoswitch an accurate diagnosis probe for mapping MMPs and evaluating the tumor development both in vitro and in vivo.
Introduction
Although there have been significant developments in conventional cancer treatment options recently, the treatments are still far from ideal due to ineffective diagnosis. It is therefore important to develop new methods for early, accurate detection and the prediction of the prognosis.1,2 Screening various biomarkers of cancers has become a significant pursuit in recent years, used in the understanding of the nature of specific cancers and improving the treatment outcomes.3 Among biomarkers, proteinases such as matrix metalloproteases (MMPs), have been proved to be closely associated with cancer progression.MMPs are a family of endopeptidases and play an important role in a series of normal physiological processes, but their expression level is typically very low.4–6 However, MMPs are increasingly expressed in various types of cancers, including breast,7 colorectal,8,9 prostate,10 and gastric cancer.11 Elevated levels of MMPs have been confirmed to be closely related with tumor proliferation, metastasis, and angiogenesis.12,13 Therefore, the recognition of MMPs as a disease biomarker has stimulated the development of imaginative strategies.
Noninvasive visualization of MMPs activity plays an important role in the early stage diagnosis of tumors. Different imaging modalities, such as magnetic resonance imaging (MRI),14–16 positron emission tomography (PET),17 and optical imaging18,19 have been developed in recent years to detect the protease activity with promising results. Among them, optical imaging based on fluorescence “turn-on” nanoprobes20 has been widely applied to detect membrane type-1 matrix metalloproteinase (MT1-MMP)-expressing tumors,21,22 colon tumors,23 human fibrosarcoma tumors24,25 and colorectal cancer.26 As opposed to “turn-on” fluorescent sensors based on mechanisms of cleaving or unfolding DNA molecules27,28 and competitive displacement and binding with Förster resonance energy transfer (FRET) acceptors,29 a MMP-sensitive nanoswitch would directly and accurately reflect in vivo distribution and activity of MMPs, as well as tumor development.
Gold nanoparticles (GNPs)30,31 have emerged as attractive probes for biosensors due to their controllable shape and size, versatile surface modification capabilities, and tunable physicochemical properties.32–34 Furthermore, GNPs have become one of the commonly used energy acceptors in several “turn-on” fluorescent probes based on FRET process35–38 to detect ions,39–41 small molecules,42 and enzymes.43 An ultrasensitive nanoprobe, based on DNA-peptide immobilized GNPs, has been reported to assess the activity of MMP-2 at concentrations as low as 10 pM with in 4 h.44 Gold nanoparticles/nanocages and their peptide-modified conjugates have also been shown to be a simple and low-cost sensing system, which could serve as a new detection method for MMPs.45–47
However, research on the assay of MMPs activity is mainly concentrated on signal enzymes (such as MMP-2 or MMP-9) in vitro presently, with no reports concerning the level of compound enzymes in tumor tissues. However, compound enzymes coexist in tumor tissues and contribute to MMPs activity, therefore investigating the activity of these compound enzymes is of great significance. In this work, we designed a fluorescent “off–on” nanoswitch (Fig. 1) for the detection of MMPs in vitro and in vivo. In this system, FITC and GNPs were connected via a peptide containing the core substrate sequence (PLGVR) of both MMP-2 and MMP-9. Acting as a response element, the peptide-modified GNPs display sensitive fluorescence response to collagenase IV, a MMP subfamily that includes MMP-2, MMP-4, and MMP-9. Thiolated polyethylene glycol monomethyl ether (SH-mPEG) plays the role of prolonging the circulation of the system in blood as well as avoiding opsonization. The nanoprobe has been well characterized by UV-Vis, fluorescence, transmission electron microscopy (TEM), and dynamitic light scattering (DLS) technology. The optimized system had a simple preparation process, the complete fluorescence quenching, and the most effective fluorescence recovery (about 50-fold intensity) when compared to reported MMP-responsive systems.16,24,26,47 In order to make the results in vitro and in vivo comparable, levels of compound enzyme in tumor cells and tumor-bearing animals were measured by ELISA method. The enzymatic distribution, depending on the level and activity of MMPs, was accurately monitored by the nanoswitch both in vitro HepG2 cells and in vivo Heps tumor-bearing mice. The biosafety and tumor penetration of the nanoswitch were also evaluated according to its cytotoxicity on HepG2 cells and accumulation in normal organs and tumor tissues.
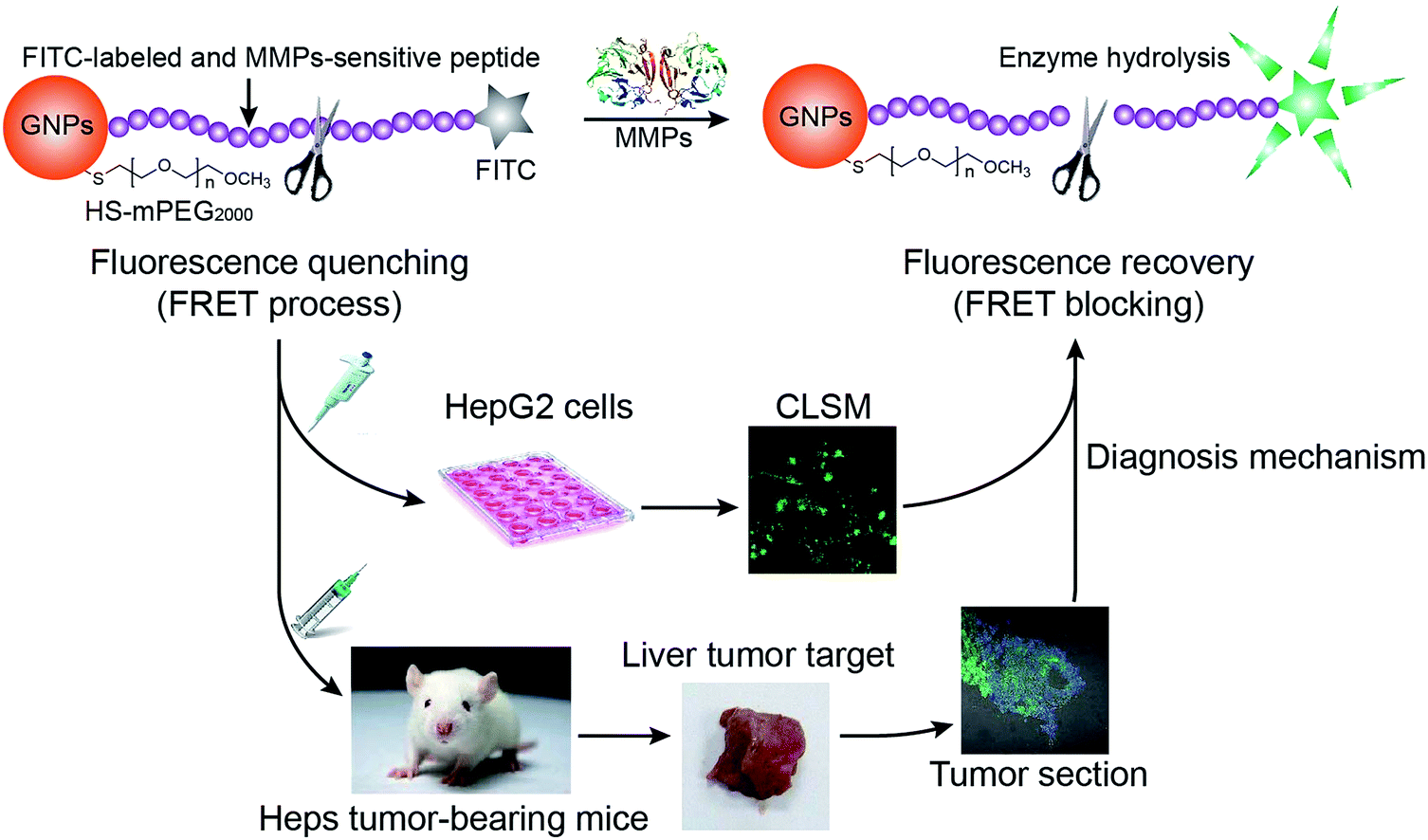 | ||
| Fig. 1 Schematic illustration of gold nanoparticle-based nanoswitch with fluorescent “off–on” function in response to matrix metalloproteinases. | ||
Results and discussion
Design, preparation, and optimization of nanoswitch
The fluorescence tranquillization of the nanoswitch is based on the FRET process.48 This process requires a non-zero integral of the spectral overlap between a photo-excited donor emission and an acceptor absorption, and the distance between the donor and the acceptor needs to be within the energy transfer distance (<10 nm).49,50 Therefore, to achieve a high transfer efficiency, a photo-excited donor (FITC here) with a good spectral overlap with its acceptor (GNPs) has been chosen to construct the system. The UV-Vis absorption spectra of GNPs and the fluorescence emission spectra of a fluorescein isothiocyanate labeled C-terminus peptide (p-FITC) are shown in Fig. 2A. The aqueous suspension of GNPs displayed a characteristic plasmon absorption at λmax = 514 nm (black line) and the aqueous solution of p-FITC showed a maximum emission at 517 nm (blue line). The almost completely overlapped spectral integral between p-FITC donor emission and GNPs acceptor absorption should guarantee an efficient FRET process and the minimum background signal of the system. | ||
Fig. 2 The preparation of GNP-p-FITC conjugate: (A) absorption spectrum (normalized) of GNPs (black line) and emission spectrum (normalized) of p-FITC (blue line). (B) Fluorescence intensity of GNP-p-FITC with the same amount of p-FITC (60 ng mL−1) but different concentrations of GNPs (3.92 × 10−5 to 98.0 μg mL−1). (C) Fluorescence intensity of GNP-p-FITC with the same amount of GNPs (39.20 μg mL−1) but different concentrations of p-FITC (1–90 ng mL−1). (D) Fluorescence intensity of GNP-p-FITC with the same ratio of GNPs to p-FITC (39.20 μg mL−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 ng mL−1) but different solution concentrations of feeding materials (GNPs: 27.44–50.96 μg mL−1; p-FITC: 35–65 ng mL−1). :50 ng mL−1) but different solution concentrations of feeding materials (GNPs: 27.44–50.96 μg mL−1; p-FITC: 35–65 ng mL−1). | ||
A peptide of 15 amino acids, containing the core substrate sequence of collagenase IV (PLGVR), was designed and introduced between GNPs and FITC to restrict the distance between GNPs and FITC in the range of 10 nm and achieve MMP-responsive fluorescence recovery. Simply, p-FITC was conjugated to the surface of GNPs through the free thiol group on the side chain of cysteine. Upon reacting with collagenase IV, the quenched fluorescence can be recovered through cleavage of the peptide (Fig. 1) at its preferential cleavage site between glycine (Gly) and valine (Val).16,25,47
In order to optimize the preparation of the system, the concentrations and optimal ratio between p-FITC and GNPs were studied in detail to ensure the lowest background signal was achieved. Fig. 2B shows the fluorescence intensity of GNP-p-FITC conjugate with a constant amount of p-FITC (60 ng mL−1) but varying concentrations of GNPs (3.92 × 10−5 to 98.0 μg mL−1). As shown in Fig. 2B, when the concentration of GNPs reaches 39.20 μg mL−1, almost all the fluorescence is quenched. A GNPs concentration of 39.20 μg mL−1 was used to optimize the amount of p-FITC. Fig. 2C shows the fluorescence intensity of GNP-p-FITC conjugate with a constant amount of GNPs (39.20 μg mL−1) but different concentrations of p-FITC (1–90 ng mL−1). When the concentration of p-FITC is 50 ng mL−1, the fluorescence intensity shows a minimum. Thus, the ratio of GNPs (39.20 μg mL−1) to p-FITC (50 ng mL−1) is considered as the optimal one for preparing the GNP-p-FITC conjugate. To further optimize the concentration for preparing GNP-p-FITC conjugate, different concentrations of GNPs and p-FITC with the same optimal ratio were studied (Fig. 2D). The concentrations were in the range of 27.44 to 50.96 μg mL−1 for GNPs and of 35 to 65 ng mL−1 for p-FITC. The results show that the concentrations of p-FITC (40 ng mL−1) and GNPs (31.36 μg mL−1) display the minimum intensities of fluorescence. We used these two concentrations in the following preparation of gold conjugates.
In this system, HS-mPEG was also conjugated to the surface of GNPs to stabilize the nanoparticles, minimize non-specific enzyme activity and reduce clearance by immunological cells,51,52 and finally to prolong the circulation time in vivo. The final product is denoted as GNP-p-FITC/mPEG. To evaluate the significance of mPEG modification on gold conjugates, the GNP-p-FITC and GNP-p-FITC/mPEG conjugates were dissolved in the PBS at different pH values (0.03 M, pH 7.4, 6.8, and 5.5), at a higher ionic strength (0.2 M, pH 7.4), and in the presence of serum (0.03 M, pH 7.4 plus 2% serum) for 12 h. As shown in Fig. S1,† before adding collagenase IV, the fluorescence intensity only slightly increases after incubation at 37 °C for 12 h in all conditions, demonstrating that the pH value, ionic strength, and serum have little influence on the fluorescence intensity change. However, there appeared to be some precipitant after 12 h of incubation in the GNP-p-FITC solution while the GNP-p-FITC/mPEG solution was still clear. This indicates that the GNP-p-FITC/mPEG conjugate shows higher stability under various conditions, especially in the presence of serum. Moreover, after adding collagenase IV for 5 min, the fluorescence intensity of the two conjugates increased significantly in all samples, with or without 12 h of incubation, suggesting sensitive fluorescence responses of the two conjugates to collagenase IV. The GNP-p-FITC/mPEG conjugate was chosen as the nanoswitch in the following experiments mainly due to its high stability in the serum-containing medium, which should improve the circulation in blood and subsequent accumulation in solid tumors.
Characterization of GNP-p-FITC/mPEG conjugate
The GNP-p-FITC/mPEG conjugate was synthesized following the procedure as described above and characterized by UV-Vis absorption spectra, fluorescence emission spectra, and TEM analysis. The UV-Vis absorption spectrum (Fig. 3A) shows that p-FITC does not exhibit significant absorbance from 200 to 800 nm. The GNPs display a characteristic UV-Vis absorption spectrum with a surface-localized plasmon resonance band at 516 nm. The GNP-p-FITC/mPEG conjugate has a similar UV absorption spectrum to GNPs, with the plasmon absorption peak at λmax = 523 nm, which shows a 7 nm red shift compared to that of GNPs demonstrating the successful modification of the p-FITC via a S–Au covalent bond. Fig. 3B shows that the GNPs do not exhibit fluorescence emission signals from 500 to 600 nm and that the FITC-labeled peptide exhibits a strong fluorescence emission at 517 nm. The prepared GNP-p-FITC/mPEG conjugate results in almost 100% quenching of the fluorescence, indicating that GNPs linked to FITC-labeled peptide can quench the fluorescence efficiently. The morphology of the prepared GNPs and GNP-p-FITC/mPEG conjugate were imaged by TEM (Fig. 3C and D), which showed that both GNPs and the conjugate had a rather uniform spherical shape and the particles were well dispersed. The DLS data shows an average diameter of ca. 19.66 ± 0.24 nm (GNPs), a little smaller than that of the GNP-p-FITC/mPEG conjugate (32.24 ± 0.62 nm) (Fig. 3E). The zeta potential value changes from −36.52 ± 1.17 mV (GNPs) to −9.99 ± 0.18 mV (GNP-p-FITC/mPEG), confirming that the citrate salt has been replaced from the surface of the GNPs by p-FITC and HS-mPEG successfully. In addition, after the incubation with physiological medium (0.03 M PBS at pH 7.4 + 2% serum here) for 2 h or 8 h, the hydrodynamic diameter of GNP-p-FITC/mPEG increased to above 200 nm (Fig. 3E), demonstrating the protein corona effect arising from the serum protein in the incubation solution.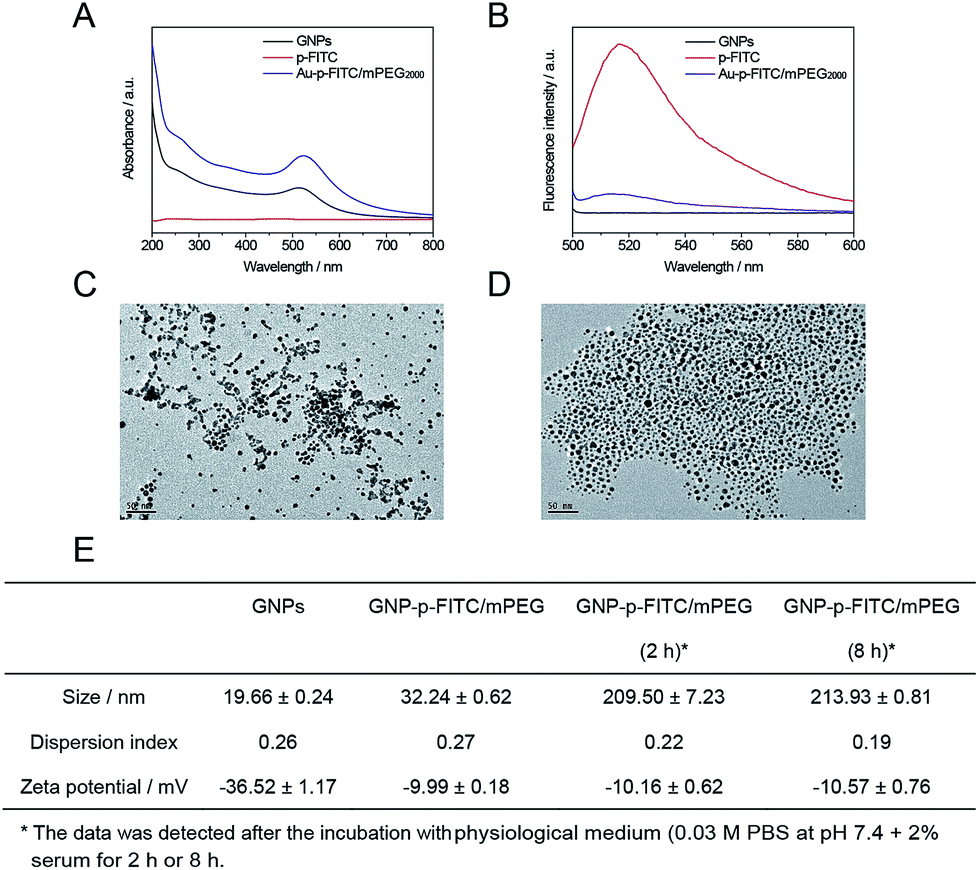 | ||
| Fig. 3 Characterizations: (A) absorption spectrum (normalized) of GNPs, p-FITC, and GNP-p-FITC/mPEG conjugate. (B) Fluorescence intensity of GNPs, p-FITC, and GNP-p-FITC/mPEG conjugate. TEM images of (C) GNPs and (D) GNP-p-FITC/mPEG conjugate. (E) Particle sizes and zeta potentials of GNPs and GNP-p-FITC/mPEG before and after the incubation with physiological medium (0.03 M PBS at pH 7.4 + 2% serum) for 2 h or 8 h, respectively, measured by the DLS instrument. | ||
Detection of enzyme levels in cell and animal and in vitro enzyme digestion experiment
In order to simulate enzyme digestion in vitro, the collagenase IV concentration in cells and animals was firstly evaluated using a mouse collagenase IV ELISA kit. As shown in Fig. 4A, the levels of collagenase IV in extracellular and intracellular Heps cells, serum (n) and plasma (n) of normal mice, serum (t) and plasma (t) of Heps tumor-bearing mice, tumor tissues of Heps tumor-bearing mice, and commercial collagenase IV were detected. The intracellular concentration of collagenase IV was slightly higher than the extracellular concentration for Heps cells, which suggests that collagenase IV is generated inside the cells, and then secreted into the extracellular matrix. In addition, collagenase IV expression in plasma and serum for Heps-bearing tumor mice was higher than the result for normal mice, although the difference is not significant. This phenomenon demonstrates the possibilities and difficulties in distinguishing the tumor using the plasma and serum samples before the metastasis.26 However, it is noted that the concentration of collagenase IV in tumor tissues is 2.63 ± 0.10 μg g−1, which is much higher than the collagenase IV concentration in cells, serum, and plasma. Moreover, using an ELASA kit to assess the commercial enzyme activity, the collagenase IV showed similar absorbance to the actual enzyme concentration in tumor tissues when its concentration was 2.4 mg mL−1. As a result, this concentration was used as a reference to investigate the enzyme cutting experiment in vitro.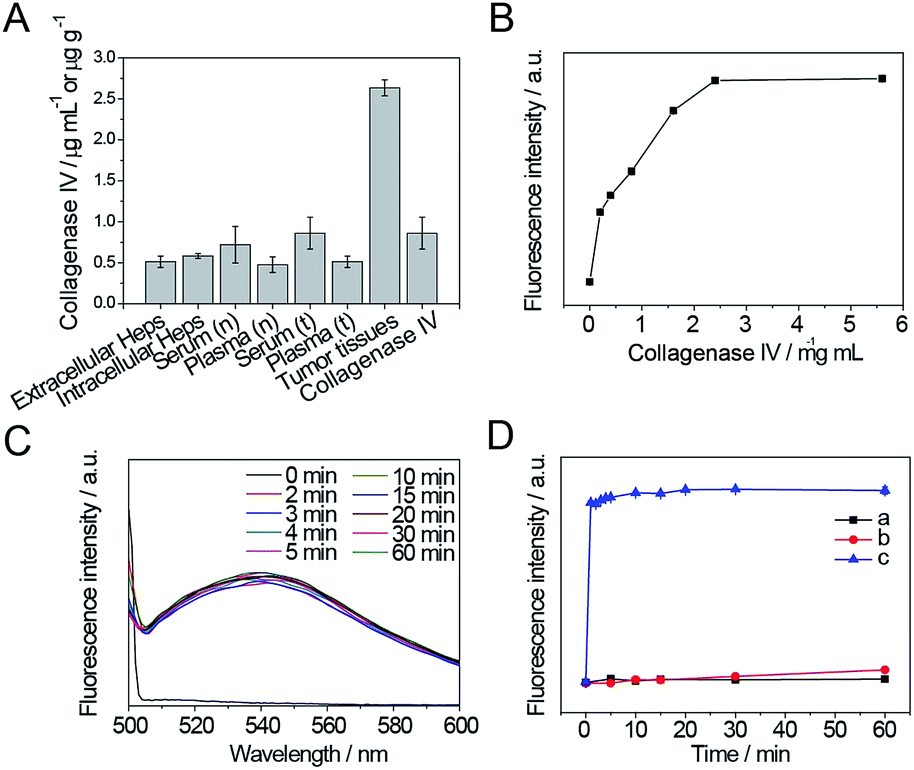 | ||
| Fig. 4 (A) Collagenase IV concentration detection by ELISA method in extracellular and intracellular Heps cells, serum (n) and plasma (n) of normal mice, serum (t) and plasma (t) of Heps tumor-bearing mice, tumor tissues and commercial collagenase IV. (B) Increase in fluorescence intensity when GNP-p-FITC/mPEG conjugate (containing 10 ng mL−1 p-FITC) is incubated at 37 °C with different amounts (from 0–5.6 mg mL−1) of collagenase IV for 15 min. (C) Fluorescence spectra of GNP-p-FITC/mPEG in the presence of 2.4 mg mL−1 of collagenase IV at different enzymatic reaction times and a temperature of 37 °C. (D) Increase in fluorescence intensity when GNP-p-FITC/mPEG conjugate (containing 10 ng mL−1 p-FITC) was incubated with (a) 2 μM GSH, (b) 10 mM GSH, and (c) 2.4 mg mL−1 collagenase IV as a function of time. | ||
The results of in vitro enzyme digestion experiments were also showed in Fig. 4. As shown in Fig. 4B, the change in fluorescence intensity of GNP-p-FITC/mPEG conjugate upon adding different amounts of collagenase IV (0–5.6 mg mL−1) was monitored to optimize the concentration of collagenase IV. The fluorescence intensity increases gradually while increasing the concentration of collagenase IV and then levels off when the concentration is higher than 2.4 mg mL−1. The enzyme reaction results in a 50-fold fluorescence enhancement. This enhancement is ascribed to the specific cleavage of the substrate by collagenase IV and subsequent FRET block via the release of the FITC-containing peptide segment from the GNPs surface. Considering the collagenase IV concentration in tumor tissues, we chose this concentration (2.4 mg mL−1) as the optimal amount of collagenase IV to be used to investigate the optimal enzymatic reaction time in the following experiments.
As shown in Fig. 4C, the fluorescence signal is fully recovered after adding collagenase IV for 2 min, and does not change in the following 1 h, which suggests a rapid enzyme cutting rate. To ensure complete enzyme catalysis for the peptide cleavage and the convenience of detection, a reaction time of 5 min is adopted.
Moreover, to confirm that the fluorescence enhancement is caused by collagenase IV rather than by the replacement of GSH, the fluorescence intensity of GNP-p-FITC/mPEG conjugate in the presence of 2 μM GSH (the extracellular GSH concentration), 10 mM GSH (the intracellular GSH concentration), and 2.4 mg mL−1 collagenase IV as a function of time was monitored. As shown in Fig. 4D, after adding collagenase IV (curve c), the fluorescence intensity is recovered, increases rapidly within the first 5 min, and then tends to level off with the reaction time. On the contrary, the fluorescence intensity of GNP-p-FITC/mPEG conjugate does not show any obvious changes when incubated with 2 μM GSH (curve a). There is a slight, yet insignificant, increase when incubated with higher a GSH concentration (10 mM, curve b) in 1 h. These results demonstrate a sensitive response of the nanoswitch toward MMPs. The fluorescence recovery mechanism is shown in Fig. 1.
Cytotoxicity assay and cellular uptake studies
Fig. 5A shows the viability of HepG2 cells after treatment with GNPs and GNP-p-FITC/mPEG conjugate. The GNP-p-FITC/mPEG conjugate (IC50 = 2.22 μg mL−1 of gold) shows a higher cytotoxicity than that of GNPs, especially at high concentrations. This phenomenon may be due to the surface modification of peptides, which enhance cellular uptake of gold conjugates.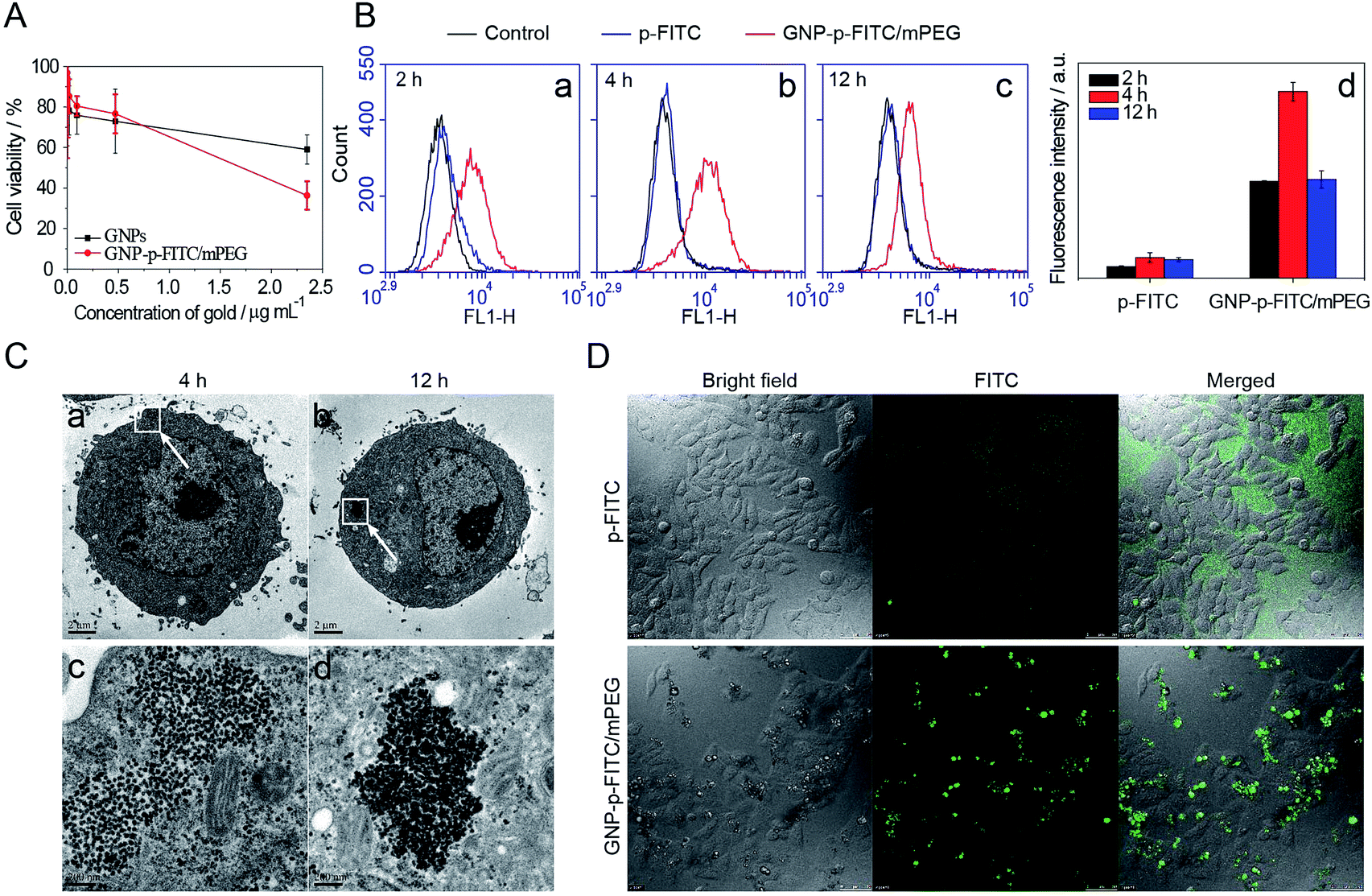 | ||
| Fig. 5 (A) Cell viability of GNPs and GNP-p-FITC/mPEG conjugate on HepG2 cells. (B) Flow cytometry assay of p-FITC and GNP-p-FITC/mPEG conjugate on HepG2 cells at (a) 2 h, (b) 4 h, and (c) 12 h, and (d) fluorescence intensity detected by flow cytometry. (C) TEM images of cells with the incubation of GNP-p-FITC/mPEG conjugate inside HepG2 cells at (a) 4 h and (b) 12 h; images (c) and (d) were the magnified images of (a) and (b), respectively. (D) Intracellular and extracellular fluorescence of p-FITC and GNP-p-FITC/mPEG conjugate on HepG2 cells after 2 h incubation (containing 5 μg mL−1 p-FITC). The fluorescence of the cells was imaged with a confocal laser scanning microscope (CLSM, Leica TCS SP5, Germany). | ||
The cellular uptake and fluorescence intensity of free p-FITC and GNP-p-FITC/mPEG conjugate at the cell level were comprehensively studied by flow cytometry, TEM, and confocal microscopy. The free p-FITC group, which displayed both the location and amount of FITC chromophore, was used as a comparison, and showed no relationship with enzyme activity. The intracellular fluorescence intensity of GNP-p-FITC/mPEG group reflects not only the cellular uptake behavior of conjugates but also the MMP activity in cells. The cells treated with only culture medium are used as a control. Fig. 5B (images a–c) shows the flow cytometry results of free p-FITC and GNP-p-FITC/mPEG conjugate, at a p-FITC dose of 5 μg mL−1, in HepG2 cells at different time intervals (2 h, 4 h, and 12 h). There is no notable difference between the control and free p-FITC in the first two hours, and the fluorescence of p-FITC does not change with time, suggesting that free p-FITC is not effectively internalized into cells. The fluorescence recovery inside cells for the GNP-p-FITC/mPEG group was 5–9 times higher compared to that of the free p-FITC at all time points (Fig. 5B, graph d), indicating the generation of MMPs inside HepG2 cells, the cellular internalization of the nanoswitch, and the dissociation of FITC from gold surface by MMPs. After 12 h of incubation, the fluorescence intensity of GNP-p-FITC/mPEG conjugate decreased, which may be due to either fluorescence quenching or the efflux of the released FITC segment.
The endocytosis of GNP-p-FITC/mPEG conjugate by HepG2 cells was further evaluated by TEM. Fig. 5C shows the morphology of HepG2 cells treated with GNP-p-FITC/mPEG and incubated for 4 h and 12 h. The particles of GNP-p-FITC/mPEG exist in the cytoplasm of HepG2 cells after 4 h of incubation. After twelve hours of incubation, the particles were attached to each other tightly, possibly due to the replacement of SH-mPEG by GSH.
Cellular imaging of free p-FITC and GNP-p-FITC/mPEG conjugate with HepG2 cells was also detected by confocal microscopy (Fig. 5D). The HepG2 cells were treated with free p-FITC and GNP-p-FITC/mPEG at a p-FITC dose of 5 μg mL−1, and incubated at 37 °C for 2 h. The fluorescence of free p-FITC is uniformly dispersed in cell culture medium but not inside cells, while the fluorescence of GNP-p-FITC/mPEG group displays bright green spots inside cells. This demonstrates that the internalization ability of p-FITC is enhanced with the help of GNPs via the endocytosis mechanism. This result is consistent with the flow cytometry data shown in Fig. 5B. Furthermore, the cellular morphology does not significantly change after treatment with free p-FITC and GNP-p-FITC/mPEG conjugate, demonstrating their low cytotoxicity.
Biodistribution of p-FITC and GNP-p-FITC/mPEG conjugate
The above studies have proved that the internalization ability of GNP-p-FITC/mPEG is much higher than that of free p-FITC. To further evaluate their accumulation in tumors and investigate their distribution in other normal organs, including heart, liver, spleen, lung and kidney, ICR male mice, with a tumor volume of 100 mm3 after Heps tumor inoculation, were intravenous injected with free p-FITC and GNP-p-FITC/mPEG conjugate at a p-FITC dose of 10 mg kg−1. Fig. 6 illustrates the fluorescence images of the tumor 12 h after administration of free p-FITC and GNP-p-FITC/mPEG conjugate using a CLSM method.53 The images show that the fluorescence of free p-FITC is distributed mainly at the margin, rather than in tumor center. This phenomenon may be a result of the osmotic pressure54 in tumor center, which prevents more p-FITC from being taken in. On the contrary, for GNP-p-FITC/mPEG conjugate, fluorescence signals were dispersed and made the tumor margin bright, which indicates that significant GNP-p-FITC/mPEG conjugate effectively entered and reached the tumor region via the EPR effect. In the tumor center, the fluorescence displayed bright green spots, which implies the successful fluorescence recovery of GNP-p-FITC/mPEG triggered by MMPs that have accumulated in tumor cells. The aforementioned data suggests that our nanoswitch can reach, accumulate at, and deeply penetrate a liver tumor, confirming its proteolytic activity in vivo.55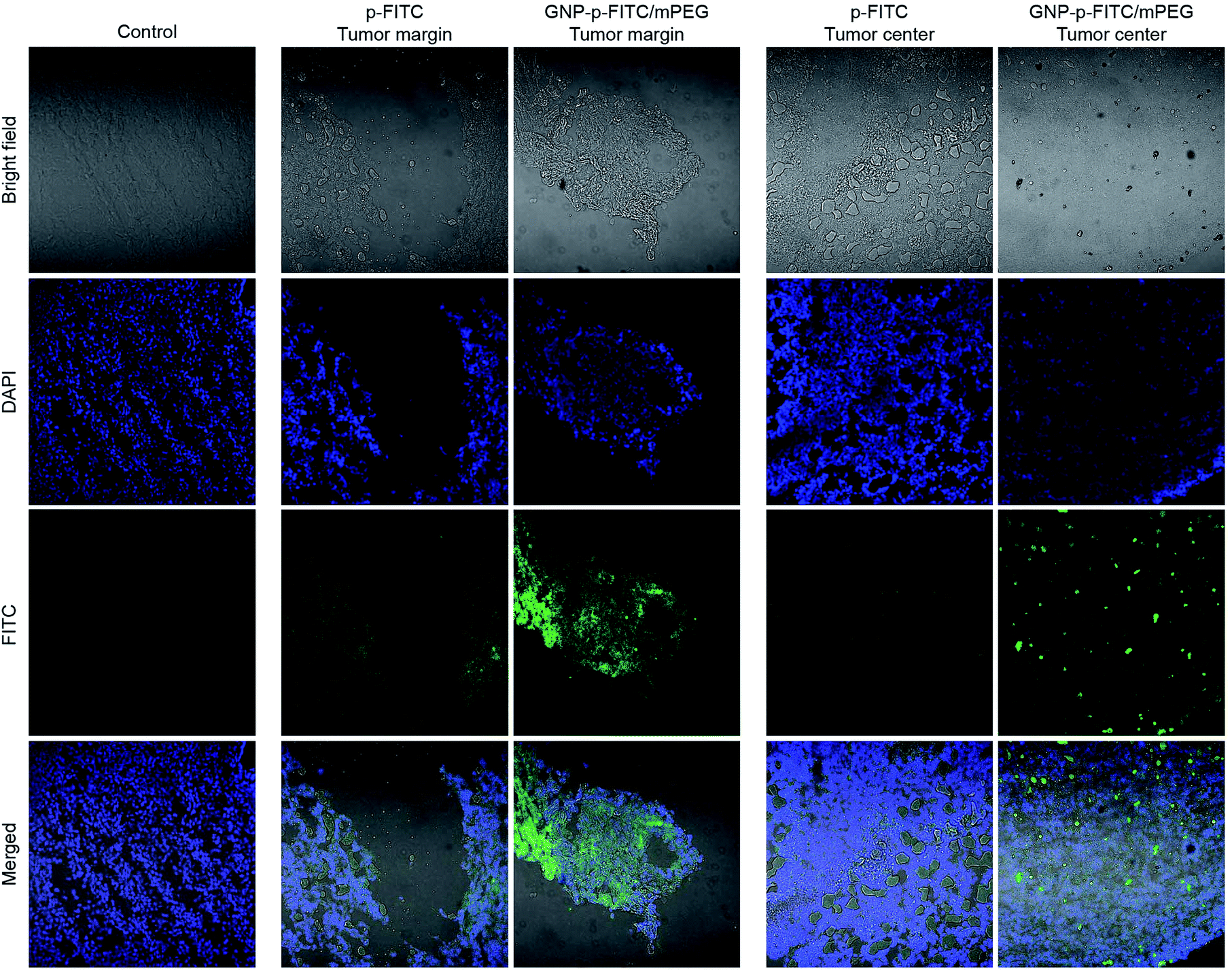 | ||
| Fig. 6 Tumor accumulations of fluorescence probes: p-FITC and GNP-p-FITC/mPEG conjugate were intravenous administrated into Heps tumor-bearing mice, the test solution at a p-FITC dose of 10 mg kg−1 for 12 h. The tumor tissue sections were prepared using Leica CM1900 Cryostat microtome (GMI, Inc, Germany) and the images were taken by confocal laser scanning microscope (CLSM, Nikon C2 plus, Japan). | ||
The biosafety and possible metabolic pathways of the nanoswitch were also studied. The distribution of free p-FITC and GNP-p-FITC/mPEG conjugate in other normal organs (including heart, liver, spleen, lung, and kidney) after 12 h of intravenous administration is shown in Fig. 7A and B, respectively. The fluorescence signal of GNP-p-FITC/mPEG conjugate was much lower than that of free p-FITC in heart, spleen and lung. This suggests that the nanoswitch may exhibit lower distribution and subsequently lower tissue toxicity in these normal tissues. It is noted that the fluorescence intensity of GNP-p-FITC/mPEG group in the liver is comparable to that of free p-FITC, demonstrating the reorganization and clearance effect of macrophages and the immune system in mice. In addition, the stronger fluorescence intensity of GNP-p-FITC/mPEG group in the kidney, compared to that of the p-FITC group, indicates a possible metabolism mechanism for FITC-containing peptide segments via the kidney.
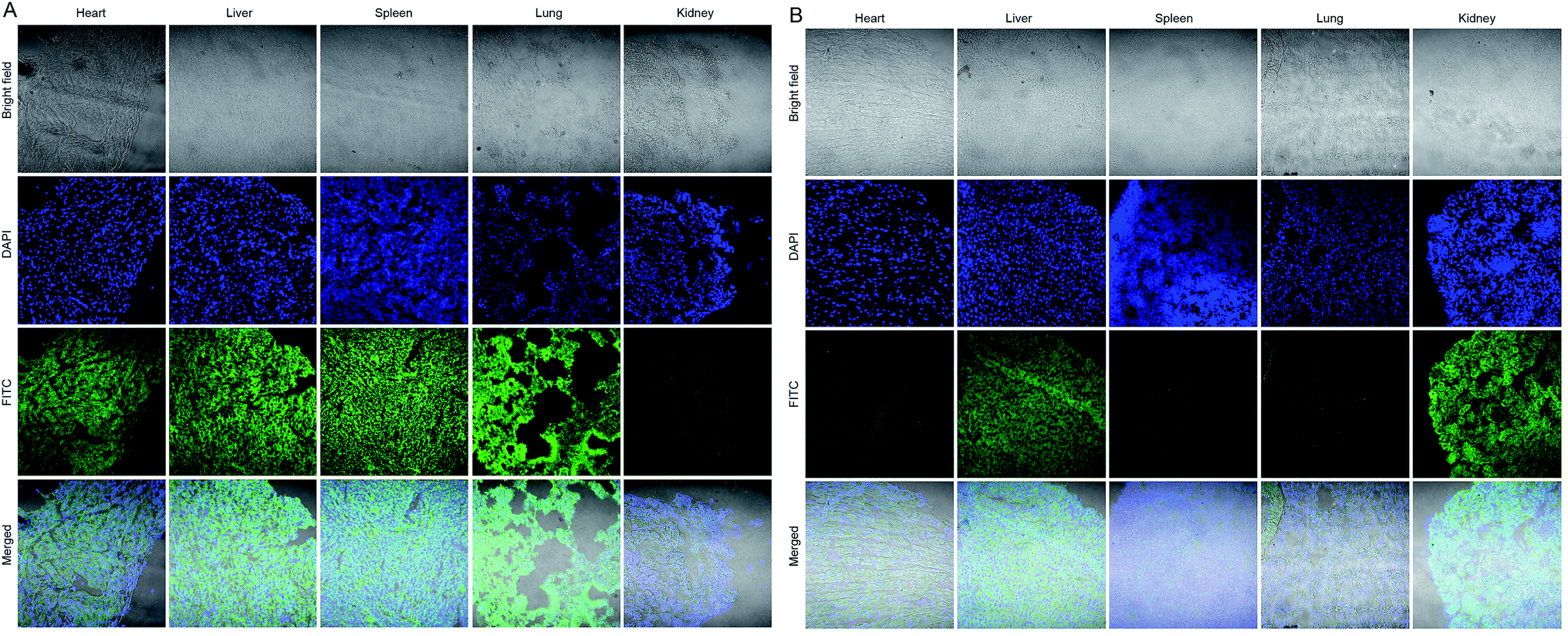 | ||
| Fig. 7 Biodistribution of (A) p-FITC and (B) GNP-p-FITC/mPEG conjugate in normal tissues, including heart, liver, spleen, lung, and kidney of Heps tumor-bearing mice after intravenous administration of test samples at a p-FITC dose of 10 mg kg−1 for 12 h. The normal tissue sections were prepared using a Leica CM1900 Cryostat microtome (GMI, Inc, Germany) and the images were taken using a confocal laser scanning microscope (CLSM, Nikon C2 plus, Japan). | ||
Materials and methods
Material and apparatus
Hydrogen tetrachloroaurate trihydrate (HAuCl4·3H2O) and thiolated polyethylene glycol monomethyl ether (HS-mPEG, molecular weight of 2000 Da) were obtained from Shanghai Chemical Reagent Company (Shanghai, China). FITC-labeled peptide (GGGGPLGVRGGGGKC-FITC) was provided by Ziyu Biotechnology Co., Ltd (Shanghai, China). Dulbecco's modified Eagle's medium (DMEM) was purchased from Gbicol (USA). 4′,6-Diamidino-2-phenylindole (DAPI) was purchased from Beyotime Institute of Biotechnology (Shanghai, China). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-tetrazoliumbromide (MTT) was purchased from Aladdin Industrial Corporation (Shanghai, China). Collagenase IV and fetal bovine serum (FBS) were purchased from Nanjing Shengxing Biotechnology Co., Ltd (Nanjing, China). Unless otherwise stated, all stating materials were obtained from commercial suppliers and were used without further purification. All aqueous solutions were prepared using deionized water (>18 MΩ, Purelab Classic Corp., USA).Fluorescence measurements were performed on an Agilent Cary Eclipse spectrophotometer (USA). UV-Vis spectra were measured with a UV-2401 PC UV/Vis spectrophotometer (Shimadzu, Japan). The particle sizes and zeta potentials were measured by DLS method with a Zetasizer zetaPALS/90 plus instrument (Brookhaven Instruments, New York, USA). Morphology of the particles was imaged using a transmission electron microscope (JEM-200CX TEM, JEOL) with an acceleration voltage of 200 kV.
Preparation process and optimization of GNP-p-FITC conjugate
GNPs with an average diameter of about 3 nm were synthesized in a single-phase system56,57 and the detail description is presented in ESI.† To monitor the preparation method of gold conjugates and achieve the optimal fluorescence quenching efficiency, the following conditions were tested: (i) keeping the amount of p-FITC constant and changing the concentration of GNP solution; (ii) keeping the amount of GNPs constant and changing the concentration of p-FITC solution; (iii) keeping the ratio of GNPs to p-FITC constant and changing the feeding amount of mixed solution. A detailed description is presented in ESI.†The preparation process of GNP-p-FITC and GNP-p-FITC/mPEG conjugates is as follows: 2.4 mL of p-FITC stock solution was mixed with 16 mL of GNPs stock solution, followed by adding deionized H2O to make the total volume of the mixed solution 60 mL. The final concentrations of p-FITC and GNPs were 40 ng mL−1 and 31.36 μg mL−1, respectively. The mixture was stirred for 0.5 h to obtain the GNP-p-FITC conjugate and then divided into two portions. One portion (30 mL) was stirred for another 1 h. 4 mg of HS-mPEG was added to the other 30 mL of the as-prepared GNP-p-FITC solution, and stirred for 1 h. The resultant GNP-p-FITC and GNP-p-FITC/mPEG solutions were dialyzed in fresh deionized water using a dialysis membrane (MWCO 7000) overnight and lyophilized. The prepared GNP-p-FITC and GNP-p-FITC/mPEG conjugates were stored in a 4 °C refrigerator for further use.
Influence of mPEG modification on the stability and fluorescence recovery of gold nanoswitch
For stability analysis, GNP-p-FITC and GNP-p-FITC/mPEG conjugate samples containing 30 ng p-FITC were dispersed in 3 mL of various buffer solutions, including 0.03 M PBS at pH 7.4, 6.8, and pH 5.5 (different pH values), 0.2 M PBS at pH 7.4 (higher ionic strength), and 0.03 M PBS at pH 7.4 with 2% serum (in the presence of serum). After that, the samples were incubated at 37 °C for 12 h to observe the clarity and fluorescence change of the conjugate solutions.For fluorescence recovery evaluation, the above-mentioned samples were added with collagenase IV (2.4 mg mL−1). Following 5 min of incubation, the fluorescence intensity of the conjugates at 518 nm was measured for the samples after 0 and 12 h of incubation in the media used for the above stability studies.
Enzymatic hydrolysis experiments
To optimize the conditions of the enzymatic hydrolysis reaction in vitro, the concentration of collagenase IV in different samples, including extracellular and intracellular Heps cells, serum (n) and plasma (n) of normal mice, serum (t) and plasma (t) of Heps tumor-bearing mice, tumor tissues, as well as commercial collagenase IV were quantified by a mouse collagenase IV ELISA kit, purchased from Shanghai Haling Biological Technology Co., Ltd (Shanghai, China), following the manufacturer's protocol.After that, the enzyme concentration and enzymatic time were investigated. In addition, to exclude the fluorescence recovery induced by the ligand reaction of glutathione (GSH), the control reaction was carried out with concentrations of GSH corresponding to extracellular or intracellular conditions for comparison. The detailed description can be found in ESI.†
Cell culture and animals
Heps cells were kindly provided by Nanjing University. Human hepatocellular liver carcinoma cell line (HepG2) was purchased from the Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China). For the cytotoxicity assay, cells were seeded in a 96-well plate at a density of 1 × 104 cells per well using DMEM with 10% FBS as a culture medium. The plates were allowed to attach at 37 °C in a humidified atmosphere with 5% CO2 for 24 h until they reached 80% confluence.ICR male mice (20 ± 2 g) were purchased from Jiesijie experimental animal Co. Ltd. (Shanghai, China, certificate number: SCXK (Hu) 2014-0006) and received care in compliance with the guidelines outlined in the Guide for the Care and Use of Laboratory Animals. The procedures were approved by the University of Science and Technology of China Animal Care and Use Committee.
Cytotoxicity assay
The cytotoxicity was assessed using a MTT assay with HepG2 cells. HepG2 cells were incubated in a growth medium with 3% FBS containing different concentrations of GNPs and GNP-p-FITC/mPEG conjugate at 37 °C in a humidified atmosphere with 5% CO2 for 4 h. The cells were then washed twice with 200 μL of PBS and incubated in a growth medium containing 1 mg mL−1 MTT for an additional 4 h at 37 °C. The medium was aspirated, and the MTT–formazan generated by living cells was dissolved in 150 μL of DMSO. The absorbance at 490 nm was measured in each well using a microplate reader (Bio-Rad). The cell viability (%) was determined by comparing the absorbance at 490 nm with control wells containing only cell culture medium. Data are presented as the average ± SD (n = 5). The concentration of GNP-p-FITC/mPEG conjugate that inhibited 50% cell growth (IC50), compared with untreated cells, was defined by curve fitting (LOGIT method) using SPSS software.Cellular uptake studies
HepG2 cells used for uptake were seeded in 6-well plates at a density of 8 × 105 cells per well and incubated at 37 °C in a humidified atmosphere with 5% CO2 overnight to reach 80% confluence. The cells were then incubated for an additional 2 h, 4 h or 12 h with p-FITC and GNP-p-FITC/mPEG conjugates at a p-FITC dose of 5 μg mL−1. Meanwhile, cells treated with a culture medium were used as a control. After incubation, excess media were removed, and the cells were washed with PBS (0.01 M, pH = 7.4), trypsinized and centrifuged. The cell pellets were resuspended in 500 μL of PBS (0.01 M, pH = 7.4). The mean fluorescence intensity of p-FITC in cells was measured by flow cytometry analysis (Accuri C6, BD, USA).For TEM tests, the cell pellets were fixed in a PBS (0.01 M, pH = 7.4) solution containing 2.5% glutaraldehyde for 4 h. Then, after being rinsed twice with a PBS solution (0.01 M, pH = 7.4), the cells were post-fixed in 1% osmic acid, dehydrated through an acetone series and embedded in Epon812 Araldite resin (polymerization at 60 °C for 15 h). Thin sections (60–80 nm) containing the cells were placed on grids and stained for 1 min with 4% uranyl acetate (1:1, acetone/water) and 0.2% Reynold's lead citrate (water). The grids were air-dried and imaged with an 80 kV JEM-1011 transmission electron microscope.
For confocal imaging tests, HepG2 cells were seeded in a confocal dish at a density of 5 × 105 cells per well and incubated overnight to reach 60% confluence. The cells were then incubated at 37 °C for an additional 2 h with p-FITC and GNP-p-FITC/mPEG conjugate at a p-FITC dose of 5 μg mL−1. The fluorescence of the cells was imaged with a confocal laser scanning microscope (CLSM, Leica TCS SP5, Germany).
Biodistribution of GNP-p-FITC/mPEG conjugate versus p-FITC
ICR male mice (20 ± 2 g) with a tumor volume of 100 mm3 (after Heps tumor inoculation) were randomly divided into two groups: (1) p-FITC and (2) GNP-p-FITC/mPEG conjugate. Each group (n = 3) was injected intravenously through the tail vein at a p-FITC dose of 10 mg kg−1. In addition, the mice treated with saline were used as a control. After administration, the mice were then sacrificed, and main organs (heart, liver, spleen, lung and kidney) and tumor tissues were collected and stored at −80 °C until they could be analyzed. All the tissue sections were prepared using a Leica CM1900 Cryostat microtome (GMI, Inc, Germany) and stained with DAPI, which is used to identify the cell nucleus. Finally, the sections were examined with a confocal laser scanning microscope (CLSM, Nikon C2 plus, Japan).Statistical analysis
All the experiments were repeated at least three times. Data are shown as the means ± standard deviation (SD). Student's test was used to determine the difference. P values less than 0.05 were considered statistically significant.Conclusions
We developed a fluorescent “off–on” nanoswitch that can be successfully used for the detection of compound enzyme MMPs activity in vitro and in vivo. This nanoswitch can be efficiently hydrolyzed by MMPs, which enables the visualization of MMPs activity in HepG2 cells and tumor-bearing mice. The detected signal displays an outstanding high signal-to-noise ratio, with almost 100% quenching of the fluorescence, and a 50-fold fluorescence enhancement in its reaction with collagenase IV in vitro. With the help of GNPs, the nanoswitch demonstrated an enhanced cellular uptake ability in HepG2 cells and a deeper penetration into the tumor issue in Heps tumor-bearing mice, due to the endocytosis and EPR effects of nanoparticles. Furthermore, lower distribution in normal tissues, compared to p-FITC, indicated lower tissue toxicity of the nanoswitch. These results demonstrate that the developed gold nanoparticle-conjugated FITC-labeled peptide system could be a promising probe for the diagnosis of human liver tumor.Acknowledgements
This work was financially supported by the Natural Science Foundation of China (31470916, 31500769), the Fundamental Research Funds for the Central Universities (2015PT036, 2016PT014), A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions, and the Open Project Program of MOE Key Laboratory of Drug Quality Control and Pharmacovigilance (DQCP2015MS01).Notes and references
- M. E. Gindy and P. K. Prud'homme, Expert Opin. Drug Delivery, 2009, 6, 865 CrossRef CAS PubMed.
- J. Xu, L. Zhao, X. Xu, N. Bertrand, W. I. Choi, B. Yameen, J. Shi, V. Shah, M. Mulvale, J. L. MacLean and O. C. Farokhzad, Angew. Chem., Int. Ed., 2015, 54, 9218 CrossRef PubMed.
- A. G. Georgakilas, Mutat. Res., 2011, 711, 1 CrossRef CAS PubMed.
- S. Zucker, M. Hymowitz, C. Conner, H. M. Zarrabi, A. N. Hurewitz, L. Matrisian, D. Boyd, G. Nicolson and S. Montana, Ann. N. Y. Acad. Sci., 1999, 878, 212 CrossRef CAS PubMed.
- P. R. Maxwell, P. M. Timms, S. Chandran and D. Gordon, Diabetic Med., 2001, 18, 777 CrossRef CAS PubMed.
- F. A. DeLano and G. W. Schmid-Schonbein, Hypertension, 2008, 52, 415 CrossRef CAS PubMed.
- A. G. Remacle, A. Noel, C. Duggan, E. MaDermott, N. O'Higgins, J. M. Foidart and M. J. Duffy, Br. J. Cancer, 1998, 77, 926 CrossRef CAS PubMed.
- E. A. Baker, F. G. Bergin and D. J. Leaper, Br. J. Surg., 2000, 87, 1215 CrossRef CAS PubMed.
- E. A. Baker and D. J. Leaper, Eur. J. Surg. Oncol., 2002, 28, 24 CrossRef CAS PubMed.
- H. Kuniyasu, P. Troncoso, D. Johnston, C. D. Bucana, E. Tahara, I. J. Fidler and C. A. Pettaway, Clin. Cancer Res., 2000, 6, 2295 CAS.
- C. F. Sier, F. J. Kubben, S. Ganesh, M. M. Heerding, G. Griffioen, R. Hanemaaijer, J. H. van Krieken, C. B. Lamers and H. W. Verspaget, Br. J. Cancer, 1996, 74, 413 CrossRef CAS PubMed.
- Z. S. Galis and J. Khatri, Circ. Res., 2002, 90, 251 CAS.
- J. Yang, Z. H. Zhang, J. Q. Lin, J. L. Lu, B. F. Liu, S. Q. Zeng and Q. M. Luo, Biochim. Biophys. Acta, 2007, 1773, 400 CrossRef CAS PubMed.
- C. Ansari, G. A. Tikhomirov, S. H. Hong, R. A. Falconer, P. M. Loadman, J. H. Grill, R. Castaneda, F. K. Hazard, L. Tong, O. D. Lenkov, D. W. Flesher, J. Rao and H. E. Daldrup-Link, Small, 2014, 10, 566 CrossRef CAS PubMed.
- J. Gallo, N. Kamaly, I. Lavadas, E. Stevens, Q. D. Nguyen, M. Wylezinska-Arridge, E. O. Aboagye and N. J. Long, Angew. Chem., Int. Ed., 2014, 53, 9550 CrossRef CAS PubMed.
- X. Y. Yue, Z. Wang, L. Zhu, Y. Wang, C. Q. Qian, Y. Ma, D. O. Kiesewetter, G. Niu and X. Y. Chen, Mol. Pharmaceutics, 2014, 11, 4208 CrossRef CAS PubMed.
- C. W. Huang, Z. Li and P. S. Conti, Bioconjugate Chem., 2012, 23, 2159 CrossRef CAS PubMed.
- Y. Cai, L. Zhu, F. Zhang, G. Niu, S. Lee, S. Kimura and X. Chen, Mol. Pharmaceutics, 2013, 10, 2237 CrossRef CAS PubMed.
- L. Zhu, F. Zhang, Y. Ma, G. Liu, K. Kim, X. Fang, S. Lee and X. Chen, Mol. Pharmaceutics, 2011, 8, 2331 CrossRef CAS PubMed.
- Y. Shimizu, T. Temma, I. Hara, A. Makino, N. Kondo, E. Ozeki, M. Ono and H. Saji, Cancer Sci., 2014, 105, 1056 CrossRef CAS PubMed.
- S. M. van Duijnhoven, M. S. Robillard, S. Hermann, M. T. Kuhlmann, M. Schäfers, K. Nicolay and H. Grüll, Mol. Pharmaceutics, 2014, 11, 1415 CrossRef CAS PubMed.
- S. M. van Duijnhoven, M. S. Robillard, K. Nicolay and H. J. Grüll, Nucl. Med., 2011, 52, 279 CrossRef CAS PubMed.
- S. M. Yoon, S. Myung, B. D. Ye, I. Kim, N. G. Lee, Y. M. Ryu, K. Park, K. Kim, I. C. Kwon, Y. S. Park, C. S. Park, D. H. Moon, D. H. Kim, M. Y. Do, J. Byeon, S. Yang and J. Kim, Gut Liver, 2010, 4, 488 CrossRef CAS PubMed.
- W. J. Akers, B. G. Xu, H. Lee, G. P. Sudlow, G. B. Fields, S. Achilefu and W. B. Edwards, Bioconjugate Chem., 2012, 23, 656 CrossRef CAS PubMed.
- X. Zhang, J. Bresee, P. P. Cheney, B. G. Xu, M. Bhowmick, M. Cudic, G. B. Fields and W. B. Edwards, Molecules, 2014, 19, 8571 CrossRef PubMed.
- Z. Wang, X. H. Li, D. Feng, L. H. Li, W. Shi and H. M. Ma, Anal. Chem., 2014, 86, 7719 CrossRef CAS PubMed.
- A. E. Prigodich, D. S. Seferos, M. D. Massich, D. A. Giljohann, B. C. Lane and C. A. Mirkin, ACS Nano, 2009, 3, 2147 CrossRef CAS PubMed.
- D. W. Huang, C. G. Niu, X. Y. Wang, X. X. Lv and G. M. Zeng, Anal. Chem., 2013, 85, 1164 CrossRef CAS PubMed.
- X. Wang and X. Q. Guo, Analyst, 2009, 134, 1348 RSC.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef CAS PubMed.
- N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547 CrossRef CAS PubMed.
- Z. Krpetic, S. Saleemi, I. A. Prior, V. See, R. Qureshi and M. Brust, ACS Nano, 2011, 5, 5195 CrossRef CAS PubMed.
- X. Xu, W. L. Daniel, W. Wei and C. A. Mirkin, Small, 2010, 6, 623 CrossRef CAS PubMed.
- D. L. Chamberland, A. Agarwal, N. Kotov, J. B. Fowlkes, P. L. Carson and X. Wang, Nanotechnology, 2008, 19, 095101 CrossRef PubMed.
- H. Lee, K. Lee, L. K. Kim and T. G. Park, Adv. Funct. Mater., 2009, 19, 1884 CrossRef CAS.
- E. Oh, D. Lee, Y. P. Kim, S. Y. Cha, D. B. Oh, H. A. Kang, J. Kim and H. S. Kim, Angew. Chem., Int. Ed., 2006, 45, 7959 CrossRef CAS PubMed.
- P. V. Kamat, S. Barazzouk and S. Hotchandani, Angew. Chem., Int. Ed., 2002, 41, 2764 CrossRef CAS.
- C. H. Fan, S. Wang, J. W. Hong, G. C. Bazan, K. W. Plaxco and A. J. Heeger, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6297 CrossRef CAS PubMed.
- S. C. Wei, P. H. Hsu, Y. F. Lee, Y. W. Lin and C. C. Huang, ACS Appl. Mater. Interfaces, 2012, 4, 2652 CAS.
- M. Li, X. J. Zhou, S. W. Guo and N. Q. Wu, Biosens. Bioelectron., 2013, 43, 69 CrossRef CAS PubMed.
- D. W. Huang, C. G. Niu, X. Y. Wang, X. X. Lv and G. M. Zeng, Anal. Chem., 2013, 85, 1164 CrossRef CAS PubMed.
- S. H. Radwan and H. M. Azzazy, Expert Rev. Mol. Diagn., 2009, 9, 511 CrossRef CAS PubMed.
- W. Gao, L. Ji, L. Li, G. Cui, K. Xu, P. Li and B. Tang, Biomaterials, 2012, 33, 3710 CrossRef CAS PubMed.
- J. H. Kim and B. H. Chung, Small, 2010, 6, 126 CrossRef CAS PubMed.
- Y. Hong, M. Ku, D. Heo, S. Hwang, E. Lee, J. Park, J. Choi, H. J. Lee, M. Seo, E. J. Lee, J. I. Yook, S. Haam, Y. Huh, D. S. Yoon, J. Suh and J. Yang, Biosens. Bioelectron., 2014, 57, 171 CrossRef CAS PubMed.
- S. Peng, Q. Zheng, X. Zhang, L. H. Dai, J. X. Zhu, Y. B. Pi, X. Q. Hu, W. Q. Cheng, C. Y. Zhou, Y. L. Sha and Y. F. Ao, ACS Appl. Mater. Interfaces, 2013, 5, 6089 CAS.
- X. H. Xia, M. X. Yang, L. K. Oetjen, Y. Zhang, Q. G. Li, J. Y. Chen and Y. N. Xia, Nanoscale, 2011, 3, 950 RSC.
- Y. M. Chen, T. L. Cheng and W. L. Tseng, Analyst, 2009, 134, 2106 RSC.
- C. S. Yun, A. Javier, T. Jennings, F. Fisher, S. Hira, S. Peterson, B. Hopkins, N. O. Reich and G. F. Strouse, J. Am. Chem. Soc., 2005, 127, 3115 CrossRef CAS PubMed.
- H. Szmacinski, K. Ray and J. R. Lakowicz, Biophotonics, 2009, 2, 243 CrossRef CAS PubMed.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288 CrossRef CAS PubMed.
- S. H. Cha, J. Hong, M. McGuffie, B. Yeom, J. S. VanEpps and N. A. Kotov, ACS Nano, 2015, 9, 9097 CrossRef CAS PubMed.
- J. C. Waters, J. Cell Biol., 2009, 185, 1135 CrossRef CAS PubMed.
- Q. Sun, X. Sun and X. Ma, Adv. Mater., 2014, 26, 7615 CrossRef CAS PubMed.
- Y. Wang, J. H. Bahng, Q. Che, J. Han and N. A. Kotov, ACS Nano, 2015, 9, 8231 CrossRef CAS PubMed.
- Y. Ding, Y. Y. Zhou, H. Chen, D. D. Geng, D. Y. Wu, J. Hong, W. B. Shen, T. J. Hang and C. Zhang, Biomaterials, 2013, 34, 10217 CrossRef CAS PubMed.
- Y. Ding, G. Gu and X. H. Xia, J. Mater. Chem., 2009, 19, 795 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Methods for preparation of GNPs, optimization process of nanoswitch, and parameters investigation of enzymatic hydrolysis, as well as the results of influence of mPEG modification on the stability and fluorescence recovery of nanoswitch (Fig. S1). See DOI: 10.1039/c5ra26172d |
| ‡ The first two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |