
Graphene oxide-based composite hydrogels with self-assembled macroporous structures†
Abstract
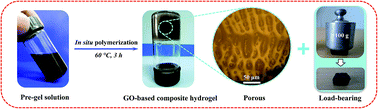
The self-assembly technique provides a new and simple route for designing porous hydrogels. At present, most of the studies in graphene oxide (GO)–polymer hydrogels are concentrated on mechanical reinforcement. Developing a self-assembled GO-based porous hydrogel along with swelling and mechanical merits is still challenging, yet very interesting and desirable for practical applications. Herein, we report self-assembled GO-based macroporous composite hydrogels by integrating GO sheets and chitosan-based hydrogel networks. GO sheets, containing adequate hydrophilic functional groups, can be dispersed well and thereby they form self-assembled supramolecular structures with polymer chains by effective intermolecular interactions (e.g., hydrogen bonding, electrostatic attraction or covalent bonding). Surprisingly, an extremely low amount (0.05–0.30 wt%) of GO can remarkably affect the architecture of hydrogel networks, leading to the formation of macroporous composite hydrogels. On the whole, the GO-based polymer composite hydrogels possess both macroporous structures (10–100 μm) and enhanced mechanical performance, yet can still retain the similar swelling properties of their parent polymeric hydrogel. Therefore, this study provides a simple route for fabricating porous hydrogels, which could find some potential applications in wastewater treatment or biomedical engineering.
 Please wait while we load your content...
Please wait while we load your content...

