
Efforts to total synthesis of philinopside E: convergent synthesis of the sulfated lanostane-type tetraglycoside†
Shujin Bai,
Zhiyong Wu,
Qingyun Huang,
Li Zhang,
Pengwei Chen,
Cong Wang,
Xiuli Zhang,
Peng Wang* and
Ming Li*
Key Laboratory of Marine Medicine, Chinese Ministry of Education, School of Medicine and Pharmacy, Ocean University of China, 5 Yushan Road, Qingdao, Shandong 266003, China. E-mail: pengw@ouc.edu.cn; lmsnouc@ouc.edu.cn
First published on 5th January 2016
Abstract
As an important step to total synthesis of philinopside E with important antitumor activities (Ed50 0.75–3.50 μg mL−1), we described herein convergent synthesis of a triterpene glycoside composed of the sulfated tetrasaccharide residue identical to that of philinopside E and the aglycone of lanost-7-en-3β-ol. The stereocontrolled synthesis of the aglycone from 24,25-dihydrolanosterol was accomplished relying on the stereoselective reductions of the 2,3-unsaturated-1,4-diketone system assisted by a C3-tert-butyldimethylsilyloxy group and convenient installation of the required 7(8)-double bond via syn elimination of triflate. Sequencial convergent coupling of monoglycoside, prepared by reacting the aglycone with orthogonally protected xylosyl thioglycoside, with trisaccharide thioglycoside originated from glucose, xylose and quinovose derivatives, incorporation of sulfation and deprotection afforded the target molecule. The features of our work are that the four 1,2-trans glycosidic bonds were stereoselectively constructed and the precious aglycone was introduced in the late-stage synthesis, which would facilitate the total synthesis of philinopside E and related natural products.
Introduction
Triterpene glycosides are a large family of secondary metabolites in plants and marine organisms that have been used in traditional medicines for thousands of years.1 To date, more than 100 sea cucumber triterpene glycosides have been isolated and characterized.2 These saponins share a common aglycone skeleton, 3β,20S-dihydroxy-5α-lanostano-18,20-lactone. Their oligosaccharide moieties, being frequently sulfated, are composed of several different monosaccharide residues, including D-xylose, D-quinovose, D-glucose, 3-O-methyl-D-glucose, and in rare cases, 3-O-methyl-D-xylose.2 Most sea cucumber saponins exhibit potential therapeutic effects in the treatment of cancer, inflammation and infections.2b,c Recently, Yi and co-workers3 isolated four new sulfated saponins, philinopside A, B, E and F, from the sea cucumber Pentacta quadrangularis, which is widely distributed throughout the South China Sea (Fig. 1). As a representative of sea cucumber triterpene glycosides, philinopside E exhibits anti-angiogenesis and antitumor activities with Ed50 values ranging from 0.75–3.50 μg mL−1.3b Studies on its mode of action revealed that philinopside E interacts with the extracellular portion of the kinase insert domain-containing receptor (KDR) to block its interaction with vascular endothelial growth factor (VEGF) and the resultant downstream signaling. This mechanism is distinct from that of conventional small-molecule inhibitors which exclusively target the cytoplasmic kinase domain of KDR.4 In addition, philinopside E markedly suppresses ανβ3 integrin-driven downstream signaling.4 However, further studies were hampered by the limited availability of this natural product. Chemical synthesis has proved to be a powerful tool to make structurally complex saponins of importance in sufficient amounts,5 however, there is no report on the total synthesis of sea cucumber triterpene glycosides such as philinopside E.6 | ||
| Fig. 1 Structure of philinopside A, B, E, F and target triterpene glycoside 1. | ||
Inspection of philinopside E suggested that a few key problems including construction of sulfated oligosaccharide residue and synthesis of highly multi-functionalized aglycone that have to be solved before a successful total synthesis could be performed. Thus, as the first attempt to gain knowledge about the synthesis of philinopside E, we undertook the synthesis of an analogue of philinopside E, the lanostane-type saponin 1 consisting of the sulfated tetrasaccharide found in philinopside E and the aglycone of lanost-7-en-3β-ol (Fig. 1). We hoped that the developed protecting-group and assembly strategies for stereocontrolled formation of glycosidic bonds and the extension of sugar chain, coupled with the strategies for synthesis of lanost-7-en-3β-ol, would pave a practical way to the total synthesis of philinopside E.
Results and discussion
As shown in Fig. 1, target molecule 1 has four 1,2-trans glycosidic bonds and is sulfated at the 4-OH position of the xylose unit. Normally, the formation of 1,2-trans glycosidic linkages is facilitated by the participation of neighboring ester protecting groups such as acetate, benzoate, and pivalate,7 and sulfate groups are introduced in the penultimate step of the synthesis.8 According to these guidelines, we proposed our plan for the synthesis of saponin 1. As outlined in Scheme 1, we envisioned that 1 could be obtained by removal of 2-naphthyl methyl (Nap) in 2, subsequent sulfation of the resulting hydroxyl and global hydrolysis of all the benzoates. The intermediate 2 could be convergently constructed by coupling lanost-7-en-3-yl xyloside 3 with the trisaccharide donor 4. Monoglycoside 3, in turn, could be synthesized from lanost-7-en-3β-ol 5 as the aglycone and the orthogonally protected xylosyl donor 6. Trisaccharide 4 could be prepared by joining three monosaccharide building blocks 7, 8 and 9. Benzoate and 2-azidomethyl benzoate are chosen as the protecting groups for the C-2 hydroxyl groups of 6–9 to ensure the highly stereoselective formation of 1,2-trans glycosidic linkages. Since the extraction of the aglycone 5 from natural sources such as Dracaena cinnabari9a and Azorella trifurcata9b is difficult and there are few methods10 available to its synthesis, we therefore would prepare the aglycone 5 from 24,25-dihydrolanosterol by designing a new stereoselective route. | ||
| Scheme 1 Retrosynthetic analysis of target triterpene glycoside 1. | ||
The synthesis of saponin 1 commenced with the synthesis of 5 en route to monoglycoside 3 (Scheme 2). Thus, tert-butyldimethylsilyl (TBS) protection of the 3-OH of 24,25-dihydrolanosterol 10 (ref. 11) under the forcing conditions, followed by allylic oxidation with KMnO4 in the presence of 18-crown-6 (ref. 12) afforded the unsaturated ketone 11 in 67% yield over two steps. After that, conjugate reduction of 11 with zinc dust in refluxing acetic acid11 gave diketone 12 in an excellent 96% yield. Simultaneous reduction of two carbonyl groups in 12 by LiAlH4 led to diol 13 as the single product in 85% yield. Notably, bulky TBS is crucial for the highly stereoselective reduction of diketone 12 because the congeners of 12 with tetrahydropyranyl and acetyl groups as the protecting group of 3-OH led to a diastereomeric mixture by reducing the carbonyl groups.13 By treating 13 with acetic anhydride in pyridine with N,N-dimethyl pyridine (DMAP) as a catalyst acetate 14 was obtained with a 91% yield. The absolute stereochemistry of 14 was unambiguously confirmed by X-ray crystallographic analysis (Scheme 2).14
 | ||
| Scheme 2 Preparation of the aglycone 5. Reagents and conditions: (a) (i) TBSCl, imidazole, N,N-dimethylformamide, 45 °C, 5 h; (ii) KMnO4, 18-crown-6, CH2Cl2/H2O, 18 °C, 24 h, 67% for two steps; (b) zinc dust, AcOH, 120 °C, 2 h, 96%; (c) LiAlH4, THF, 13 °C, 3 h, 85%; (d) Ac2O, pyridine, DMAP, 17 °C, overnight, 91%; (e) MsCl, Et3N, CH2Cl2, 15 °C, 89%; (f) HF·pyridine, THF, 40 °C, 88%; (g) H2 (130 atm), Pd/C, AcOH, MeOH, 120 °C, 72 h, 95%; (h) (i) BzCl, pyridine, DMAP, 18 °C, 6 h; (ii) AcCl, CHCl3/MeOH, 40 °C, 24 h, 78% for two steps; (i) MsCl, Et3N, CH2Cl2, 13 °C, 5 h, 87%; (j) NaOAc, AcOH, 120 °C, 0.5 h, 87% (20/21 = 1/12.5); (k) Tf2O, pyridine, CH2Cl2, −10 °C, 5 h, 96% (20/21 = 1/20); (l) LiAlH4, THF, 21 °C, 4 h, 89%. | ||
On the basis of the crystal structure, the synthesis of 14 deserves some comments. The ORTEP view of the crystal illustrates that two angular methyl groups at C-10 and C-13 are both β-oriented, which can cause steric hindrance to 11β-OH on the same face of the rings. This explains why the acetylation of diol 13 preferentially occurred to the 7β-OH. It should be also pointed out that in 1H NMR spectrum of 14 the axial 7α-H resonates at 4.91 ppm as a doublet of triplets, while the equatorial 11α-H appears at 4.22 ppm as a broad singlet. These splitting patterns can be predicted by Karplus equation15 on the basis of stereochemical assignment of 14. The same trends were observed for 7α-H and 11α-H in 13.
With the access to 14 secured, we initially sought to remove the equatorial 11β-OH through a Barton–McCombie radical deoxygenation reaction16 which involves conversion of the alcohol to a xanthate, followed by reduction with tri-n-butyltin hydride in refluxing toluene. However, subjecting the alcohol 14 to the standard conditions did not afford the xanthate required for the deoxygenation step. Inspired by the work of Deslongchamps et al.,17 we then tended to use the LiAlH4 reduction of mesylate to remove the 11β-OH. Unexpectedly, treatment of alcohol 14 with mesyl chloride and triethyl amine resulted in the formation of alkene 15 (Scheme 2), which is probably caused by the elimination of the mesylate generated in situ. The ready availability of alkene 15 led to an alternative strategy for removing the 11β-OH, namely, hydrogenation of the 9(11)-double bond in 15. After many attempts, we found that the presence of the bulky TBS group at the C-3 position significantly hampered the hydrogenation reaction. This issue can be simply solved by removing the TBS group. After deprotection, alcohol 16 could undergo hydrogenation under 130 atmospheric pressures at 120 °C to provide 17 in an excellent 95% yield.
With alcohol 17 in hand, we turned our attention to the synthesis of 5 by a syn elimination (Scheme 2). Thus, benzoylation of the 3-OH followed by a facile chemoselective deacetylation using methanolic hydrogen chloride18 afforded alcohol 18. Inspired by the synthetic work of Purdy and co-workers,19 who utilized syn elimination of mesylate to form the desired double bond, mesylate 19, prepared by mesylation of 18, was treated with sodium acetate in refluxing acetic acid to furnish a mixture of olefins 20 and 21 in 87% yield with a ratio of 1/12.5. The ratio was determined by 1H NMR spectroscopy based on comparing integration values for the H-7 of 20 (5.53 ppm) and 21 (5.22 ppm). It is well established that triflate is a better leaving group than mesylate,20 and triflates have been widely used in formation of double bonds in steroids.21 Consequencely, in order to improve the regioselectivity of the elimination reaction, 18 was converted into a triflate using triflic anhydride and pyridine in CH2Cl2, which underwent rapid elimination to form 21 with an exclusive selectivity (20/21 = 1/20). This result implies that an E1 mechanism might be responsible for the elimination of triflate. Finally, reductive deprotection of benzoate with LiAlH4 supplied 5 in 89% yield, 1H data of which are identical to the reported.22 Thus the transformation of 24,25-dihydrolanosterol into 5 was achieved in 12 steps with an overall 24% yield.
With the aglycone 5 in hand, it is necessary to synthesize pyranoxylosyl donor 6 to make monoglycoside 3. Briefly, Nap protection of the 4-OH in thioglycoside 22 (ref. 23) followed by removal of isopropylidene with methanol in the presence of camphorsulfonic acid (CSA) furnished diol 23 with a 96% yield over two steps. Dibutylstannylene-mediated regioselective benzoylation24 of 23 afforded benzoate 24 in a satisfactory 60% yield. Condensation of alcohol 24 with 2-azidomethyl benzoic acid (AzmbOH) in the presence of 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDCI) gave the xylosyl thioglycoside 6. The glycosylation of 5 with 6 promoted by N-iodosuccinimide (NIS) and trimethylsilyl triflate (TMSOTf) proceeded smoothly to furnish 25 in 91% yield. Notably, this procedure left the 7(8)-double bond on 5 intact. The newly formed 1,2-trans glycosidic linkage on 25 is confirmed by the coupling constant of 3JH1′–H2′ = 7.0 Hz. In addition to allowing for the selective formation of 1,2-trans glycosidic bonds through neighboring group participation, Azmb is often used as a temporary protecting group in carbohydrate synthesis because it can be selectively removed in the presence of other acyl groups including benzoyl group (Bz).25 As expected, the Azmb group on 25 was selectively cleaved in the presence of a Bz group by first reducing the azide with Bu3P and then intramolecular aminolysis of benzoate25 to give alcohol 3 in 92% yield (Scheme 3).
 | ||
| Scheme 3 Synthesis of monoglycoside 3. Reagents and conditions: (a) (i) NapBr, NaH, N,N-dimethylformamide, 27 °C, 6 h; (ii) CSA, CH2Cl2/MeOH, 28 °C, 5 h, 96% for two steps; (b) (i) Bu2SnO, toluene, 120 °C, 2 h; (ii) BzCl, 0 °C, 5 h, 60%; (c) AzmbOH, EDCI, DMAP, CH2Cl2, 13 °C, 11 h, 91%; (d) 5, NIS, TMSOTf, CH2Cl2, 4 Å molecular sieves, 0 °C, 2 h, 91%; (e) Bu3P, H2O, THF, 50 °C, 1 h, 92%. | ||
With the synthesis of 3 fully realized, the next significant step as outlined in Scheme 1 for convergent synthesis of 1 is the extension of the sugar chain on 3 by trisaccharide donor 4. It was initially expected that thioglycoside 4 could be assembled by a reaction sequence involving coupling of xylosyl acceptor 7a with the trichloroacetimidate donor 8, transformation of the resultant disaccharide into the corresponding imidate and subsequent glycosylation with thioglycoside 9. For this purpose, 7a was synthesized by first selective acetylation26 of the 3-OH of thioglycoside 26,23 benzoylation of both 2-OH and 4-OH, and final removal of the acetyl group with methanolic hydrogen chloride.18 Glucosyl trichloroacetimidate 8 was then prepared from hemiacetal 28.27 After that, TMSOTf-promoted glycosylation of 7a with 8 was performed. Disappointingly, the reaction did not produce any of the desired disaccharide and only gave glucosyl thioglycoside 29 in 56% yield (Scheme 4 eqn (1)). From this observation, we assumed that the vicinal electron-withdrawing benzoates render the 3-OH of 7a significantly less nucleophilic than the anomeric thioether of the same molecule. Thus aglycone transfer28 of thioglycoside 7a preferentially occurred in the reaction.
 | ||
| Scheme 4 Preparation of disaccharide 34. Reagents and conditions: (a) (i) Ac2O, TBAOAc, CH3CN, 40 °C, 6 h; (ii) BzCl, pyridine, DMAP, 25 °C, 4 h, 59% for 27, 60% for 31; (b) AcCl, CH2Cl2/MeOH, 26 °C, 24 h, 95% for 7a, 93% for 7b; (c) Cl3CCN, DBU, CH2Cl2, 25 °C, 8 h, 95%; (d) TMSOTf (0.1 equiv.), 4 Å molecular sieves, CH2Cl2, 0 °C, 1 h, 56%; (e) TMSOTf (0.1 equiv.), 4 Å molecular sieves, CH2Cl2, 0 °C, 1 h 35% for 32; (f) TBSOTf (0.3 equiv.), 4 Å molecular sieves, CH2Cl2, 0 °C, 1 h, 39% for 33β, 23% for 33α; (g) TMSOTf, 4 Å molecular sieves, CH2Cl2, 0 °C, 1 h, 80%. | ||
To circumvent this unexpected aglycone migration, p-methoxyphenyl (PMP) group, being less nucleophilic than phenylthio group, was introduced as an anomeric protecting group of xylosyl acceptor. In an analogous route to that of 7a, xylosyl acceptor 7b was obtained from 30.29 Unfortunately, upon treatment of 7b with 8 in the presence of TMSOTf (0.1 equiv.), orthoester 32 was obtained in 35% yield (Scheme 4 eqn (2)). By switching the activation agent from TMSOTf to tert-butyldimethylsilyl triflate (TBSOTf) (0.3 equiv.), the reaction did produce the desired β-linked disaccharide 33β in 39% yield, but along with α-isomer 33α in 23% yield. Although it is often disfavored by the presence of a neighboring participatory substituent at the C-2 position of the glycosyl donor, the formation of 1,2-cis glycosidic linkages is not unknown in similar reactions.7,30 The formation of undesired 33α was ascribed to the decreased reactivity of 3-OH due to intramolecular hydrogen-bond between the anomeric PMP with 3-OH on 7b. 1H NMR data of 7b demonstrates that 7b partially adopts a 1C4 chair conformation, thus offering opportunities for intramolecular hydrogen-bond formation. 1H NMR studies31 and theoretical calculations32 on molecules similar to 7b lend support to those inferences.
Not satisfied with the relatively poor stereochemical outcome of the reaction of 7b with 8, we sought another approach to the desired disaccharide. Previous work by other groups33 illustrates that benzyl α-xylosides with 3-OH free are competent glycosyl acceptors in a wide variety of glycosylation events. On the basis of these results, TMSOTf-promoted coupling of benzyl α-xyloside 7c (ref. 24b) with trichloroacetimidate 8 was conducted. Happily, this reaction afforded the desirable disaccharide 34 in 80% yield (Scheme 4 eqn (3)). Conversion of benzyl glycoside 34 into disaccharide trichloroacetimidate 35 was achieved by removing the benzyl group of 34 with hydrogenolysis and subsequently treating the resultant alcohol with trichloroacetonitrile in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (Scheme 5). Gratifyingly, coupling of 35 and quinovosyl thioglycoside acceptor 9 proceeded smoothly providing the expected trisaccharide thioglycoside 4 in 70% yield. Thioglycoside 9, in turn, could be readily prepared by selective iodination of primary hydroxyl of 36 (ref. 34) using Ph3P/I2/imidazole and the following deiodination of the resulting 37 by hydrogenolysis.
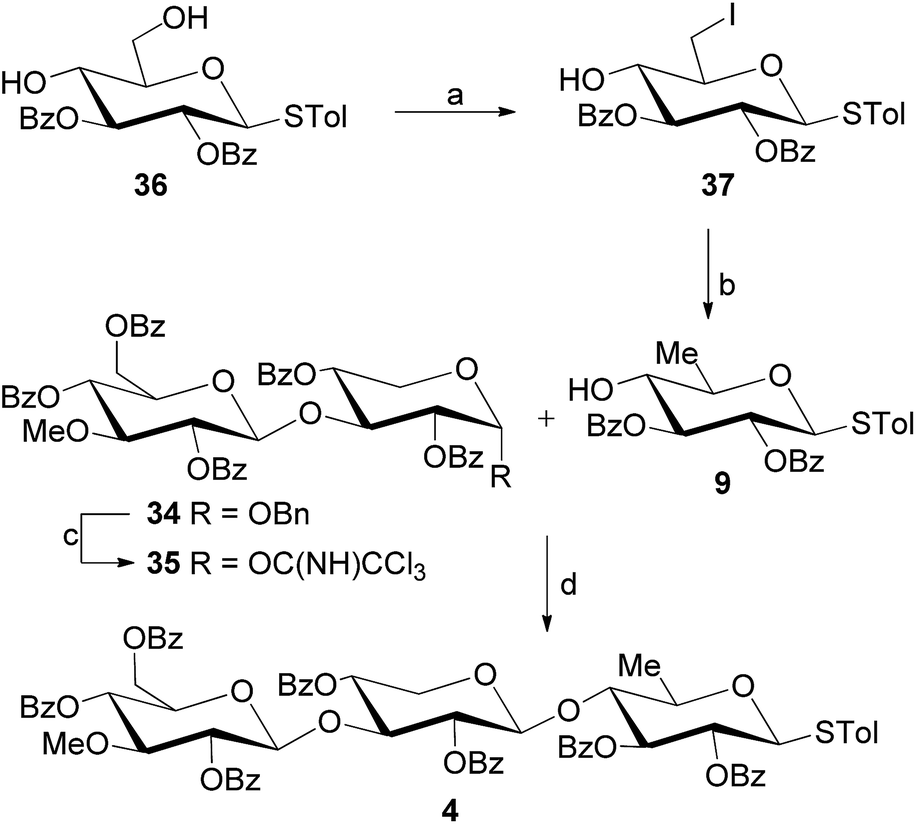 | ||
| Scheme 5 Synthesis of trisaccharide 4. Reagents and conditions: (a) PPh3, imidazole, I2, THF, 29 °C, 1 h; (b) H2, 10% Pd–C, NaHCO3, N,N-dimethylformamide, 40 °C, 24 h, 83%; (c) (i) H2, 10% Pd/C, EtOH, 50 °C, 24 h; (ii) Cl3CCN, DBU, CH2Cl2, 5 h, 74% for two steps; (d) TMSOTf, 4 Å molecular sieves, CH2Cl2, 0 °C, 1 h, 70%. | ||
With key two building blocks 3 and 4 available, the synthesis of our target molecule 1 was in sight. As shown in Scheme 6, NIS-promoted reaction of 3 and 4 in the presence of TMSOTf gave rise to the tetrasaccharide saponin 2 in a satisfactory 67% yield. Oxidative removal of the Nap group was then effected by 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) in MeOH/CH2Cl2 to give alcohol 38 in 88% yield. Using pyridine·SO3 as the sulfating agent, sulfated product 39 was then obtained in an excellent 94% yield by microwave-assisted sulfation35 of 38 in pyridine at 100 °C. After treatment of 39 with MeONa in methanol to globally deprotect benzoyl groups and subsequent dialysis against water, target molecule 1 was finally obtained. 1H and 13C spectroscopic data due to the tetrasaccharide moiety of 1 match those of philinopside E.
 | ||
| Scheme 6 Synthesis of saponin 1. Reagents and conditions: (a) NIS, TMSOTf, CH2Cl2, 4 Å molecular sieves, 2 h, 0 °C, 67%; (b) DDQ, CH2Cl2/MeOH, 20 °C, 5 h, 88%; (c) pyridine·SO3, pyridine, microwave, 100 W, 100 °C, 2 h, 94%; (d) NaOMe, MeOH, 25 °C, 24 h, 86%. | ||
Conclusions
In conclusion, a tritepene saponin, comprised of the same tetrasaccharide residue as that of philinopside E and the aglycone of lanost-7-en-3β-ol, has been accomplished by a convergent coupling of trisaccharide thioglycoside donor with monoglycoside acceptor. Lanost-7-en-3β-ol was efficiently prepared from commercially available 24,25-dihydrolanosterol in a stereocontrolled manner involving stereoselective reduction of unsaturated 1,4-diketone system and facile installation of 7(8)-double bond by syn elimination of triflate. The successful access to trisaccharide thioglycoside demonstrated that a benzyl α-xyloside was a critical glycosyl acceptor for the extension of sugar chain at its 3-OH. Since the sugar fragment of philinopside E is found in more than 40 sea cucumber triterpene glycosides,2,3 the protecting-group and assembly tactics in this work, used to stereoselectively construct glycosidic linkages and to extend sugar chain, sets the foundation for the synthesis, structural modification and biological evaluation of philinopside E and its congeners.Experimental
General information
All nonaqueous reactions were carried out under an atmosphere of argon in flame- or oven-dried glassware with magnetic stirring unless otherwise indicated. Dichloromethane for glycosylation reactions was distilled from calcium hydride. All other commercially obtained reagents were used as received, except where specified otherwise. Flash column chromatography was performed on Silica Gel H (300–400 mesh, Qingdao, China). Analytical thin layer chromatography was performed on Silicycle SiliaPlate glass-backed plates coated with silica gel (60 Å pore size, F-254 indicator) and visualized by exposure to ultraviolet light and/or staining with aqueous 8% sulfuric acid in methanol. Optical rotations were determined with a digital polarimeter. High-resolution mass spectral (HRMS) data were determined with a LTQ Orbitrap. 1H and 13C NMR spectra were recorded on a 500 or 600 MHz NMR spectrometer with Me4Si as the internal standard. Chemical shifts are recorded in δ values and J values were given in Hz.3β-(tert-Butyldimethylsilyloxy)-lanost-8-en-7,11-dione (11)
To a solution of 10 (3.70 g, 8.63 mmol) in N,N-dimethylform-amide (12 mL) were added TBSCl (2.60 mL, 17.26 mmol) and imidazole (2.07 g, 34.52 mmol). After the reaction mixture was warmed at 45 °C, and stirred for 5 h, the volatile was evaporated in vacuo. The residue was taken up in CH2Cl2 and washed with 1 M HCl, saturated aqueous NaHCO3 and brine. The collected organic phase was dried over Na2SO4, filtered, and concentrated under the reduced pressure. The resultant crude product was used next step. A 100 mL round bottom flask open to atmosphere was charged with KMnO4 (1.16 g, 7.37 mmol) and 18-crown-6 (1.95 g, 7.37 mmol), followed by the addition of 30 mL of water. A solution of the obtained above product in 30 mL of CH2Cl2 was then added to the resulting suspension by a quick syringe transfer. The reaction mixture was vigorously stirred under argon at rt for 24 h, then the solid was filtered off with a Buche funnel and washed with CH2Cl2. The filtrate was separated, and the aqueous layer was extracted with CH2Cl2. The combined organic layers were dried over Na2SO4, filtered and concentrated. The residues were purified by silica gel column chromatography (petroleum ether/CH2Cl2 = 3/1) to give 11 (478 mg, 0.84 mmol, 67% for two steps). [α]24D = +56.4 (c 1.10, CHCl3); 1H NMR (500 MHz, CDCl3) δ 3.23 (dd, J = 11.4, 4.3 Hz, 1H), 2.88–2.78 (m, 1H), 2.73 (d, J = 16.0 Hz, 1H), 2.59 (d, J = 16.0 Hz, 1H), 2.56–2.40 (m, 2H), 2.21–2.07 (m, 1H), 2.03–1.90 (m, 1H), 1.29 (s, 3H), 1.16 (s, 3H), 0.93 (s, 3H), 0.91–0.84 (m, 18H), 0.84 (s, 3H), 0.80 (s, 3H), 0.03 (s, 3H), 0.02 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 202.70, 202.65, 152.2, 150.7, 78.4, 51.9, 50.4, 49.3, 49.1, 47.5, 39.9, 39.6, 39.5, 36.9, 36.4, 36.3, 34.2, 32.4, 28.4, 28.2, 28.1, 27.5, 26.02, 26.00, 24.1, 23.0, 22.7, 18.7, 18.2, 17.8, 17.0, 16.1, −3.6, −4.8; HRMS (ESI): m/z calcd for C36H63O3Si [M + H]+ 571.4541, found 571.4538.3β-(tert-Butyldimethylsilyloxy)-lanost-7,11-dione (12)
To the reflux solution of 11 (478 mg, 0.84 mmol) in a glacial acetic acid (30 mL), zinc dust (2.10 g) was added portionwise during 1 h and the stirring was continued for another 1 h. At this stage the solid was filtered off and washed with CH2Cl2. The filtrate was concentrated in vacuo. The residues were purified by silica gel column chromatography (petroleum ether/CH2Cl2 = 3/1) to give 12 (463 mg, 0.81 mmol, 96%). [α]24D = +33.4 (c 1.05, CHCl3); 1H NMR (500 MHz, CDCl3) δ 3.19 (dd, J = 11.5, 4.3 Hz, 1H), 2.85–2.74 (m, 1H), 2.63 (d, J = 13.1 Hz, 1H), 2.55 (d, J = 13.6 Hz, 1H), 2.42–2.28 (m, 3H), 2.24–2.10 (m, 2H), 2.09–1.97 (m, 1H), 1.26 (s, 3H), 1.19 (s, 3H), 0.92–0.81 (m, 21H), 0.79 (s, 3H), 0.70 (s, 3H), 0.03 (s, 3H), 0.02 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 209.8, 209.7, 79.0, 60.8, 53.2, 52.69, 52.65, 49.1, 48.8, 46.6, 40.1, 39.7, 39.6, 36.9, 36.4, 36.1, 36.0, 33.2, 28.8, 28.3, 28.1, 27.8, 26.0, 24.1, 23.0, 22.7, 18.6, 18.2, 17.7, 16.3, 15.5, 13.9, −3.7, −4.8; HRMS (ESI): m/z calcd for C36H65O3Si [M + H]+ 573.4697, found 573.4691.3β-(tert-Butyldimethylsilyloxy)-lanost-7β,11β-diol (13)
To a solution of 12 (700 mg, 1.22 mmol) in anhydrous THF (25 mL) at rt was added LiAlH4 (465 mg, 12.20 mmol). After stirring for 3 h, the reaction mixture was cooled in an ice bath and quenched by the addition of 5% HCl. The resulting solution was extracted with EtOAc. The combined organic phase was washed with brine, dried with Na2SO4, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc = 25/1) to afford compound 13 (599 mg, 1.04 mmol, 85%). [α]24D = +47.4 (c 0.70, CHCl3); 1H NMR (500 MHz, CDCl3) δ 4.20 (s, 1H), 3.72–3.55 (m, 1H), 3.19 (dd, J = 11.4, 3.6 Hz, 1H), 1.19 (s, 3H), 1.01 (s, 3H), 0.92–0.83 (m, 24H), 0.81 (s, 3H), 0.04 (s, 3H), 0.03 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 79.6, 72.8, 68.7, 52.4, 51.6, 50.4, 48.2, 45.3, 44.6, 42.7, 39.6, 39.5, 37.44, 37.40, 36.5, 36.2, 32.1, 29.0, 28.5, 28.2, 27.9, 26.1, 24.2, 23.0, 22.7, 19.0, 18.3, 18.1, 17.3, 17.1, 16.3, −3.6, −4.8; HRMS (ESI): m/z calcd for C36H68O3SiNa [M + Na]+ 599.4830, found 599.4817.7β-Acetoxy-3β-(tert-butyldimethylsilyloxy)-lanost-11-ol (14)
Diol 13 (264 mg, 0.458 mmol) was dissolved in pyridine (20 mL), then DMAP (6 mg, 0.05 mmol) and Ac2O (130 μL, 1.37 mmol) was sequentially added at 0 °C. The reaction was allowed to stir overnight under argon atmosphere. The reaction was quenched with water and extracted with EtOAc (2 × 40 mL). The combined organic phases were washed thrice with hydrochloric acid (1 M) and brine. The collected organic phase was dried over Na2SO4, filtered and concentrated under reduced pressure, which was purified by silica gel column chromatography (petroleum ether/EtOAc = 50/1) to give 14 (258 mg, 0.42 mmol, 91%). [α]24D = +46.4 (c 0.85, CHCl3); 1H NMR (500 MHz, CDCl3) δ 4.91 (td, J = 10.5, 5.8 Hz, 1H), 4.22 (s, 1H), 3.19 (dd, J = 11.4, 4.1 Hz, 1H), 2.25 (dd, J = 12.4, 10.2 Hz, 1H), 2.01 (s, 3H), 1.21 (s, 3H), 1.01 (s, 3H), 0.92–0.82 (m, 21H), 0.81 (s, 3H), 0.78 (s, 3H), 0.04 (s, 3H), 0.03 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 170.7, 79.6, 74.3, 68.6, 52.0, 51.5, 50.6, 48.1, 45.1, 42.7, 40.9, 39.61, 39.57, 37.3, 37.2, 36.7, 36.2, 35.2, 29.0, 28.4, 28.3, 28.1, 27.7, 26.1, 24.2, 23.0, 22.7, 22.1, 19.0, 18.3, 17.8, 17.3, 17.0, 16.3, −3.7, −4.8; HRMS (ESI): m/z calcd for C38H70O4SiNa [M + Na]+ 641.4936, found 641.4932.7β-Acetoxy-3β-(tert-butyldimethylsilyloxy)-lanost-9-ene (15)
To a mixture of 14 (80 mg, 0.13 mmol) and Et3N (36 μL, 0.26 mmol) in CH2Cl2 (2 mL) was added methanesulfonyl chloride (MsCl, 20 μL, 0.26 mmol) at 0 °C. After the reaction had been completed, the mixture was diluted with CH2Cl2 and washed with saturated aqueous NH4Cl and brine. The organic phase was dried over Na2SO4, filtered and concentrated under reduced pressure. The residues were purified by silica gel column chromatography (petroleum ether/EtOAc = 20/1) to give 15 (80 mg, 0.11 mmol, 89%). [α]24D = +34.6 (c 1.00, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.33 (d, J = 6.2 Hz, 1H), 4.92 (td, J = 11.0, 5.2 Hz, 1H), 3.18 (dd, J = 10.8, 4.9 Hz, 1H), 2.43 (d, J = 11.0 Hz, 1H), 2.04 (s, 3H), 1.08 (s, 3H), 0.90–0.84 (m, 21H), 0.77 (s, 3H), 0.76 (s, 3H), 0.67 (s, 3H), 0.04 (s, 3H), 0.03 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 170.5, 145.8, 118.0, 79.3, 77.2, 74.5, 50.4, 48.2, 46.7, 46.4, 45.0, 39.6, 39.0, 36.9, 36.6, 36.3, 36.2, 35.8, 28.61, 28.57, 28.3, 28.2, 27.9, 26.1, 24.2, 23.0, 22.7, 22.1, 22.0, 18.6, 18.5, 18.3, 16.1, 14.5, −3.7, −4.8.7β-Acetoxylanost-9-ene (16)
To a solution of compound 15 (157 mg, 0.26 mmol) in THF (3.0 mL) in a TEFLON tube was added a solution of 70% HF·pyridine (0.88 mL). The reaction mixture was stirred for 6 h at 40 °C, and quenched with saturated NaHCO3 solution. The resultant mixture was extracted with EtOAc. The combined organic layers were washed with saturated aqueous NaHCO3, dried over Na2SO4, filtered and concentrated. The crude product was purified by silica gel column chromatography (petroleum ether/EtOAc = 5/1) to give 16 (112 mg, 0.23 mmol, 88%). [α]24D = +48.8 (c 0.55, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.34 (d, J = 6.1 Hz, 1H), 4.92 (td, J = 11.0, 5.2 Hz, 1H), 3.31–3.14 (m, 1H), 2.43 (d, J = 10.6 Hz, 1H), 2.04 (s, 3H), 1.08 (s, 3H), 0.99 (s, 3H), 0.90–0.83 (m, 9H), 0.81 (s, 3H), 0.77 (s, 3H), 0.67 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 170.5, 145.5, 118.3, 78.8, 77.2, 74.4, 50.4, 48.3, 46.7, 46.4, 45.0, 39.6, 39.1, 36.9, 36.6, 36.3, 36.2, 35.8, 28.6, 28.20, 28.15, 27.8, 27.7, 24.2, 23.0, 22.7, 22.1, 22.0, 18.6, 18.5, 15.7, 14.5; HRMS (ESI): m/z calcd for C32H54O3Na [M + Na]+ 509.3965, found 509.3965.7β-Acetoxy-24,25-dihydrolanosterol (17)
To a solution of olefin 16 (65 mg, 0.13 mmol) in MeOH (12 mL) was added 10% Pd/C (100 mg) and AcOH (600 μL). The reaction vessel was evacuated and backfilled with hydrogen (130 atm). The mixture was kept at 120 °C for 72 h, then filtered through a thin plug of Celite. The filtrate was washed with EtOAc (2 × 50 mL) and concentrated in vacuo. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc = 5/1) to afford 17 (62 mg, 0.13 mmol, 95%). [α]24D = +37.7 (c 0.90, CHCl3); 1H NMR (500 MHz, CDCl3) δ 4.85 (td, J = 10.6, 5.3 Hz, 1H), 3.21 (dd, J = 11.4, 3.2 Hz, 1H), 2.00 (s, 3H), 1.96–1.77 (m, 3H), 0.96 (s, 3H), 0.94 (s, 3H), 0.88–0.84 (m, 9H), 0.83 (s, 3H), 0.79 (s, 3H), 0.78 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 170.6, 78.9, 77.2, 74.6, 51.6, 50.1, 47.3, 46.9, 45.9, 43.0, 39.6, 38.9, 37.4, 36.7, 36.2, 35.5, 31.9, 28.5, 28.24, 28.16, 28.1, 27.7, 24.2, 23.0, 22.7, 22.1, 20.3, 18.9, 16.7, 15.6, 14.5, 13.9; HRMS (ESI): m/z calcd for C32H56O3Na [M + Na]+ 511.4122, found 511.4123.3β-Benzoyloxylanost-7β-ol (18)
To a solution of 17 (1.30 g, 3.03 mmol) in pyridine (20 mL) was added DMAP (37 mg, 0.30 mmol) and BzCl (528 μL, 4.55 mmol) at 0 °C. The reaction was stirred for 6 h at room temperature and quenched with water. After removal of solvent, the residue was dissolved in CH2Cl2 and washed with 0.1 M HCl, saturated aqueous NaHCO3. The organic phase was dried over anhydrous Na2SO4, filtered and concentrated. The resulting residue was dissolved in 65 mL of CH3OH/CHCl3 (v/v = 1.6/1) followed by dropwise addition of acetyl chloride (4.80 mL) at 0 °C. The reaction mixture was stirred for 24 h at 40 °C, and then quenched with Et3N. The volatile was evaporated in vacuo. The resulting residue was purified by silica gel column chromatography (petroleum ether/EtOAc = 15/1) to afford 18 (1.04 g, 1.89 mmol, 62%) with the recovery of acetate (363 mg, 0.61 mmol). [α]24D = +35.7 (c 1.00, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.04 (d, J = 7.7 Hz, 2H), 7.55 (t, J = 7.3 Hz, 1H), 7.44 (t, J = 7.6 Hz, 2H), 4.72 (dd, J = 11.4, 4.3 Hz, 1H), 3.64 (td, J = 10.0, 4.9 Hz, 1H), 1.05 (s, 3H), 1.00 (s, 3H), 0.94 (s, 3H), 0.91 (s, 3H), 0.79 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 166.4, 132.9, 131.0, 129.6, 128.4, 81.5, 77.2, 73.1, 52.1, 49.8, 47.4, 46.7, 46.4, 46.1, 39.6, 38.2, 37.2, 36.8, 36.7, 36.6, 36.2, 31.9, 31.6, 28.7, 28.3, 28.1, 24.2, 22.9, 22.7, 20.3, 18.9, 17.0, 16.8, 14.5, 14.1; HRMS (ESI): m/z calcd for C37H58O3Na [M + Na]+ 573.4278, found 573.4283.Lanost-7β-O-methylsulfony-3β-yl benzoate (19)
To a solution of 18 (100 mg, 0.18 mmol) in CH2Cl2 (2.0 mL) was added MsCl (42 μL, 0.55 mmol) and Et3N (126 μL, 0.91 mmol) dropwise at 0 °C. The reaction mixture was stirred for 5 h at room temperature, and then quenched with MeOH. The volatile was evaporated in vacuo. The resulting residue was purified by silica gel column chromatography (petroleum ether/EtOAc = 15/1) to afford 19 (99 mg, 0.16 mmol, 87%). [α]24D = +33.4 (c 0.55, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.03 (d, J = 7.3 Hz, 2H), 7.56 (t, J = 7.4 Hz, 1H), 7.44 (t, J = 7.7 Hz, 2H), 4.79 (td, J = 10.6, 5.3 Hz, 1H), 4.71 (dd, J = 11.6, 4.3 Hz, 1H), 3.01 (s, 3H), 2.41–2.26 (m, 1H), 1.05 (s, 3H), 1.02 (s, 3H), 0.95 (s, 3H), 0.94 (s, 3H), 0.90–0.84 (m, 9H), 0.79 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 166.3, 133.0, 130.9, 129.7, 128.5, 83.2, 81.2, 51.9, 50.0, 47.1, 47.0, 46.1, 44.0, 40.1, 39.6, 38.2, 37.0, 36.7, 36.5, 36.2, 35.4, 31.6, 29.6, 28.5, 28.3, 28.1, 24.2, 24.1, 23.0, 22.7, 20.4, 19.0, 17.0, 16.5, 14.5, 14.0.3β-Benzoyloxylanost-7-ene (21)
Lanost-7-en-3β-ol (5)
To a solution of 21 (700 mg, 1.31 mmol) in THF (15 mL) was added LiAlH4 (250 mg, 6.57 mmol) at 0 °C. The reaction was stirred for 4 h at room temperature and quenched with 0.1 M HCl. The mixture was extracted with ethyl acetate. The organic phase was washed with 0.1 M HCl, brine, dried over anhydrous Na2SO4, filtered and concentrated. The residue was applied to silica gel column chromatography (petroleum ether/EtOAc = 15/1) to afford 5 (501 mg, 1.17 mmol, 89%). [α]24D = +3.8 (c 0.70, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.21 (d, J = 4.3 Hz, 1H), 3.25 (dd, J = 11.0, 4.5 Hz, 1H), 0.99 (s, 3H), 0.97 (s, 3H), 0.91–0.84 (m, 15H), 0.64 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 145.1, 116.8, 79.5, 52.1, 51.0, 50.4, 47.4, 44.5, 39.7, 38.8, 38.3, 36.7, 36.6, 35.8, 32.3, 32.2, 28.4, 28.2, 27.8, 27.6, 25.0, 24.3, 23.1, 23.0, 22.7, 20.2, 19.1, 16.2, 15.6, 14.3; HRMS (ESI): m/z calcd for C30H52ONa [M + Na]+ 451.3910, found 451.3908.p-Tolyl 4-O-(2-naphthylmethyl)-1-thio-β-D-xylopyranoside (23)
Alcohol 22 (1.69 g, 5.71 mmol) was dissolved in N,N-dimethylformamide (50 mL) followed by addition of NaH (457 mg, 11.42 mmol) at 0 °C. After stirring for 20 min under argon atmosphere, NapBr (1.55 mL, 6.85 mmol) was added to the reaction mixture. With stirring for another 6 h, the reaction was quenched by methanol and poured into water. The mixture was extracted with CH2Cl2. The organic phase was collected and washed with HCl (1 M), saturated NaHCO3, dried over Na2SO4, filtered and concentrated in vacuo. The residue was dissolved in 50 mL of CH2Cl2/MeOH (v/v = 1/1), and then CSA (1.69 g, 5.71 mmol) was added. The reaction was stirred at room temperature for 5 h, then quenched by Et3N and concentrated. The residue was purified by silica gel column (petroleum ether/EtOAc = 2/1) to afford 23 (2.17 g, 5.48 mmol, 96% for two steps). [α]24D = −61.8 (c 1.00, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.87–7.72 (m, 4H), 7.52–7.35 (m, 5H), 7.11 (d, J = 7.8 Hz, 2H), 4.85 (d, J = 11.9 Hz, 1H), 4.79 (d, J = 11.9 Hz, 1H), 4.47 (d, J = 9.0 Hz, 1H), 4.09 (dd, J = 11.5, 4.8 Hz, 1H), 3.69 (t, J = 8.4 Hz, 1H), 3.56–3.45 (m, 1H), 3.35 (t, J = 8.7 Hz, 1H), 3.29 (t, J = 10.7 Hz, 1H), 2.33 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 138.6, 135.5, 133.5, 133.3, 133.2, 123.0, 128.6, 128.0, 127.8, 126.9, 126.4, 126.2, 125.8, 89.0, 76.9, 76.7, 73.3, 72.0, 67.3, 21.3; HRMS (ESI): m/z calcd for C23H28O4NS [M + NH4]+ 414.1734, found 414.1727.p-Tolyl 3-O-benzoyl-4-O-(2-naphthylmethyl)-1-thio-β-D-xylopyranoside (24)
Diol 23 (1.88 g, 4.74 mmol) and dibutyltin oxide (1.30 g, 5.22 mmol) was dissolved in anhydrous toluene (45 mL). The resulting mixture was refluxed for 2 h with a Dean–Stark trap to remove the formed water during the reaction. At this stage the reaction was cooled to room temperature followed by addition of benzoyl chloride (605 μL, 5.22 mmol) dropwise. After the mixture was stirred for another 5 h, the volatile was removed in vacuo. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc = 8/1) to afford 24 (1.42 g, 2.84 mmol, 60%). [α]24D = −90.9 (c 0.75, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.11 (d, J = 7.5 Hz, 2H), 7.84–7.76 (m, 1H), 7.76–7.67 (m, 3H), 7.58 (t, J = 7.4 Hz, 1H), 7.52–7.33 (m, 7H), 7.13 (d, J = 8.0 Hz, 2H), 5.40 (t, J = 6.2 Hz, 1H), 4.99 (d, J = 5.5 Hz, 1H), 4.84 (d, J = 12.1 Hz, 1H), 4.80 (d, J = 12.1 Hz, 1H), 4.40 (dd, J = 12.2, 3.3 Hz, 1H), 3.77 (t, J = 5.8 Hz, 1H), 3.75–3.70 (m, 1H), 3.65 (dd, J = 12.2, 6.2 Hz, 1H), 2.34 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 166.0, 138.3, 134.7, 133.5, 133.3, 133.2, 133.0, 130.2, 130.0, 129.6, 128.59, 128.56, 128.0, 127.8, 127.0, 126.4, 126.2, 125.8, 89.5, 73.7, 72.5, 70.4, 63.5, 21.3; HRMS (ESI): m/z calcd for C30H28O5SNa [M + Na]+ 523.1550, found 523.1554.p-Tolyl 2-O-[2-(azidomethyl)benzoyl]-3-O-benzoyl-4-O-(2-naphthyl-methyl)-1-thio-β-D-xylopyranoside (6)
To a solution of 24 (200 mg, 0.40 mmol) in dry CH2Cl2 (5 mL) were added AzmbOH (106 mg, 0.60 mmol), EDCI (153 mg, 0.80 mmol), and DMAP (97 mg, 0.80 mmol) under argon atmosphere. The mixture was stirred for 12 h, and diluted with CH2Cl2 followed by washing with 1 M HCl, saturated aqueous NaHCO3, and brine. The organic phase was dried over Na2SO4, filtered and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc = 7/1) to give 6 (239 mg, 0.36 mmol, 91%). [α]24D = +33.4 (c 1.00, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.98 (dd, J = 8.2, 1.0 Hz, 2H), 7.92 (dd, J = 7.8, 1.0 Hz, 1H), 7.79–7.71 (m, 1H), 7.69–7.59 (m, 3H), 7.59–7.53 (m, 1H), 7.51–7.33 (m, 8H), 7.30 (dd, J = 8.4, 1.5 Hz, 1H), 7.28–7.22 (m, 1H), 7.11 (d, J = 8.0 Hz, 2H), 5.62 (t, J = 8.1 Hz, 1H), 5.27 (t, J = 8.3 Hz, 1H), 4.95 (d, J = 8.4 Hz, 1H), 4.78 (d, J = 12.2 Hz, 1H), 4.72 (d, J = 12.2 Hz, 1H), 4.64 (d, J = 14.8 Hz, 1H), 4.54 (d, J = 14.8 Hz, 1H), 4.32 (dd, J = 11.9, 4.7 Hz, 1H), 3.90–3.76 (m, 1H), 3.58 (dd, J = 11.9, 8.9 Hz, 1H), 2.33 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 165.7, 165.3, 138.6, 137.6, 135.0, 133.5, 133.4, 133.21, 133.15, 133.0, 131.3, 130.1, 130.0, 129.41, 129.35, 128.7, 128.6, 128.4, 128.20, 128.15, 128.0, 127.8, 126.9, 126.3, 126.2, 125.8, 86.9, 74.4, 74.1, 73.0, 70.7, 66.6, 52.8, 21.3; HRMS (ESI): m/z calcd for C38H33O6N3SNa [M + Na]+ 682.1988, found 682.2000.Lanost-7-en-3β-yl 2-O-[2-(azidomethyl)benzoyl]-3-O-benzoyl-4-O-(2-naphthylmethyl)-β-D-xylopyranoside (25)
A solution of alcohol 5 (24 mg, 0.057 mmol) and thioglycoside 6 (50 mg, 0.074 mmol) in dry CH2Cl2 (2 mL) was stirred vigorously in the presence of activated 4 Å molecular sieves (200 mg) for 20 min. The mixture was cooled to 0 °C, then NIS (26 mg, 0.114 mmol) and TMSOTf (1 μL, 0.0057 mmol) were added. After stirring for 2 h, the reaction mixture was quenched with Et3N, and the solid was filtered off. The filtrate was washed with saturated aqueous Na2S2O3 and brine. The organic phase was dried over Na2SO4, filtered and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc = 17/1) to give 25 (51 mg, 0.052 mmol, 91%). [α]24D = +45.6 (c 1.00, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.94 (d, J = 7.4 Hz, 3H), 7.77–7.71 (m, 1H), 7.70–7.59 (m, 3H), 7.53 (t, J = 7.4 Hz, 1H), 7.50–7.39 (m, 4H), 7.36 (t, J = 7.8 Hz, 2H), 7.32–7.27 (m, 2H), 5.62 (t, J = 8.8 Hz, 1H), 5.30 (dd, J = 9.0, 7.2 Hz, 1H), 5.13 (d, J = 5.3 Hz, 1H), 4.77 (d, J = 12.3 Hz, 1H), 4.74–4.68 (m, 2H), 4.62 (s, 2H), 4.14 (dd, J = 11.9, 4.9 Hz, 1H), 3.90–3.80 (m, 1H), 3.50 (dd, J = 11.8, 9.5 Hz, 1H), 3.12 (dd, J = 11.7, 3.5 Hz, 1H), 0.94 (s, 3H), 0.90–0.84 (m, 9H), 0.83 (s, 3H), 0.76 (s, 3H), 0.72 (s, 3H), 0.61 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 165.9, 165.1, 145.0, 137.9, 135.1, 133.4, 133.23, 133.17, 133.0, 131.3, 130.0, 129.6, 129.2, 128.50, 128.45, 128.0, 127.9, 127.8, 127.0, 126.3, 126.2, 125.9, 116.7, 103.1, 90.2, 75.0, 73.7, 73.1, 72.2, 63.4, 52.9, 52.1, 51.0, 50.6, 47.3, 44.5, 39.7, 38.8, 38.2, 36.7, 36.6, 35.4, 32.3, 32.2, 28.15, 28.13, 27.7, 26.2, 24.9, 24.3, 23.0, 22.9, 22.7, 20.2, 19.1, 16.4, 16.2, 14.2; HRMS (ESI): m/z calcd for C61H77O7N3Na [M + Na]+ 986.5659, found 986.5671.Lanost-7-en-3β-yl 3-O-benzoyl-4-O-(2-naphthylmethyl)-β-D-xylopyranoside (3)
To a solution of 25 (400 mg, 0.41 mmol, 1 equiv.) in THF (10 mL) were added Bu3P (815 μL, 3.26 mmol, 8 equiv.) and water (0.40 mL) under argon atmosphere. The reaction mixture was stirred for 1 h at 50 °C, when TLC monitoring indicated that the reactant was completely consumed. The reaction mixture was diluted with EtOAc, washed with saturated aqueous NaHCO3 and brine. The organic phase was dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (petroleum ether/CH2Cl2/EtOAc = 20/5/1) to give 3 (300 mg, 0.38 mmol, 92%). [α]24D = −29.1 (c 1.00, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.04 (d, J = 7.4 Hz, 2H), 7.82–7.75 (m, 1H), 7.75–7.66 (m, 3H), 7.57 (t, J = 7.4 Hz, 1H), 7.50–7.34 (m, 5H), 5.37 (t, J = 7.0 Hz, 1H), 5.18 (d, J = 2.6 Hz, 1H), 4.81 (d, J = 12.2 Hz, 1H), 4.78 (d, J = 12.2 Hz, 1H), 4.60 (d, J = 5.2 Hz, 1H), 4.14 (dd, J = 12.1, 3.8 Hz, 1H), 3.78–3.71 (m, 1H), 3.71–3.65 (m, 1H), 3.51 (dd, J = 12.0, 7.2 Hz, 1H), 3.18 (dd, J = 11.7, 3.4 Hz, 1H), 2.87 (d, J = 6.3 Hz, 1H), 0.97 (s, 3H), 0.96 (s, 3H), 0.91–0.84 (m, 15H), 0.64 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 166.1, 145.1, 135.0, 133.31, 133.26, 133.2, 130.1, 129.9, 128.5, 128.4, 128.0, 127.8, 127.0, 126.3, 126.2, 125.9, 116.7, 104.7, 90.0, 74.1, 73.1, 72.6, 71.3, 61.7, 52.1, 51.0, 50.6, 47.3, 44.5, 39.7, 39.0, 38.3, 36.7, 36.6, 35.4, 32.3, 32.2, 28.5, 28.2, 27.8, 26.3, 25.0, 24.3, 23.0, 22.9, 22.7, 20.2, 19.1, 16.6, 16.2, 14.3; HRMS (ESI): m/z calcd for C53H72O6Na [M + Na]+ 827.5221, found 827.5230.p-Tolyl 3-O-acetyl-2,4-di-O-benzyl-1-thio-β-D-xylopyranoside (27)
26 (100 mg, 0.39 mmol) was allowed to react with acetic anhydride (41 μL, 0.43 mmol) in dry acetonitrile (1.5 mL) at 40 °C for 12 h in the presence of tetrabutylammonium acetate (35 mg, 0.12 mmol). The solution was concentrated in vacuo and directly purified by flash column chromatography (petroleum ether/EtOAc = 1.5/1). The resulting residue was dissolved in pyridine (2 mL) and then benzoyl chloride (111 μL, 0.93 mmol) and DMAP (3 mg, 0.023 mmol) were added. After the reaction went to completion, the mixture was concentrated in vacuo. The residue was diluted with CH2Cl2 and washed with 1 M HCl, saturated aqueous solution of NaHCO3 and brine. The combined organic phase dried over Na2SO4, filtered and concentrated. The obtained residue was purified by silica gel column chromatography (petroleum ether/EtOAc = 8/1) to afford 27 (116 mg, 0.23 mmol, 59% for two steps). [α]24D = −52.1 (c 1.00, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.03 (d, J = 7.4 Hz, 2H), 8.00 (d, J = 7.5 Hz, 2H), 7.62–7.53 (m, 2H), 7.47–7.33 (m, 6H), 7.12 (d, J = 7.9 Hz, 2H), 5.54 (t, J = 7.7 Hz, 1H), 5.26 (t, J = 7.6 Hz, 1H), 5.21–5.13 (m, 1H), 5.01 (d, J = 7.6 Hz, 1H), 4.54 (dd, J = 11.9, 4.6 Hz, 1H), 3.64 (dd, J = 11.9, 8.0 Hz, 1H), 2.34 (s, 3H), 1.97 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 169.9, 165.6, 165.2, 138.7, 133.62, 133.56, 133.54, 130.1, 130.0, 129.9, 129.4, 129.3, 128.7, 128.65, 128.60, 86.9, 71.4, 70.3, 69.2, 65.0, 21.3, 20.8; HRMS (ESI): m/z calcd for C28H30O7NS [M + NH4]+ 524.1737, found 524.1723.p-Tolyl 2,4-di-O-benzoyl-1-thio-β-D-xylopyranoside (7a)
To a solution of 27 (2.86 g, 5.65 mmol) in 50 mL of MeOH/CH2Cl2 (v/v = 1/2) was added acetyl chloride (2.0 mL) dropwise at 0 °C. The reaction mixture was stirred for 24 h, and then quenched with Et3N. The volatile was removed in vacuo. The resulting residue was purified by silica gel column chromatography (petroleum ether/EtOAc = 6/1) to afford 7a (2.58 g, 5.37 mmol, 95%). [α]24D = −59.3 (c 1.00, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.06 (d, J = 8.1 Hz, 2H), 8.02 (d, J = 8.1 Hz, 2H), 7.65–7.50 (m, 2H), 7.49–7.31 (m, 6H), 7.11 (d, J = 7.8 Hz, 2H), 5.19–5.06 (m, 2H), 5.00 (d, J = 7.5 Hz, 1H), 4.48 (dd, J = 11.9, 4.4 Hz, 1H), 4.14 (t, J = 7.5 Hz, 1H), 3.61 (dd, J = 11.8, 8.0 Hz, 1H), 2.34 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 166.3, 138.6, 133.63, 133.58, 133.52, 130.2, 130.0, 129.9, 129.57, 129.55, 128.9, 128.61, 128.59, 86.4, 73.1, 72.4, 71.7, 64.6, 21.4; HRMS (ESI): m/z calcd for C26H25O6S [M + H]+ 465.1366, found 465.1353.2,4,6-Tri-O-benzoyl-3-O-methyl-α-D-glucopyranosyl trichloroacetimidate (8)
To a solution of hemiacetal 28 (2.90 g, 5.73 mmol) in CH2Cl2 (50 mL) were added Cl3CCN (5.75 mL, 57.3 mmol) and DBU (0.17 mL, 1.15 mmol). The resulting mixture was stirred at room temperature for 6 h followed by concentration in vacuo. The residue was purified by silica gel flash column chromatography (petroleum ether/EtOAc = 7/1) to give 8 (3.53 g, 5.44 mmol, 95%). [α]24D = +81.5 (c 1.00, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.60 (s, 1H), 8.16–7.95 (m, 6H), 7.67–7.50 (m, 3H), 7.51–7.34 (m, 6H), 6.71 (d, J = 3.7 Hz, 1H), 5.58 (t, J = 9.8 Hz, 1H), 5.43 (dd, J = 9.8, 3.8 Hz, 1H), 4.59 (dd, J = 12.2, 2.4 Hz, 1H), 4.44–4.54 (m, 1H), 4.39 (dd, J = 12.2, 5.2 Hz, 1H), 4.18 (t, J = 9.6 Hz, 1H), 3.50 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 166.1, 165.3, 165.1, 160.4, 133.5, 133.5, 133.1, 129.9, 129.8, 129.7, 129.6, 129.2, 129.2, 128.5, 128.5, 128.3, 93.4, 79.0, 72.10, 70.6, 70.0, 62.7, 60.8; HRMS (ESI): m/z calcd for C30H26O9NCl3Na [M + Na]+ 672.0565, found 672.0555.p-Tolyl 2,4,6-tri-O-benzoyl-3-O-methyl-1-thio-β-D-glucopyranoside (29)
A solution of compound 7a (38 mg, 0.082 mmol) and 8 (80 mg, 0.12 mmol) in anhydrous CH2Cl2 (2 mL) was stirred vigorously in the presence of activated 4 Å molecular sieves (2 g) for 20 min. The mixture was cooled to 0 °C and TMSOTf (1.5 μL, 0.0082 mmol) was added. After 1 h, the reaction mixture was quenched with Et3N, and the solid was filtered off. The filtrate was concentrated in vacuo and the resulting residue was purified by silica gel column chromatography (petroleum ether/EtOAc = 7/1) to afford 29 (28 mg, 0.046 mmol, 56%). [α]24D = +9.4 (c 1.25, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.15–8.08 (m, 2H), 8.03 (m, 4H), 7.65–7.54 (m, 3H), 7.51–7.40 (m, 6H), 7.35 (d, J = 8.1 Hz, 2H), 6.88 (d, J = 8.0 Hz, 2H), 5.41 (t, J = 9.6 Hz, 1H), 5.28 (t, J = 9.4 Hz, 1H), 4.84 (d, J = 10.0 Hz, 1H), 4.64 (dd, J = 12.1, 2.7 Hz, 1H), 4.40 (dd, J = 12.1, 6.3 Hz, 1H), 4.05–3.96 (m, 1H), 3.86 (t, J = 9.1 Hz, 1H), 3.38 (s, 3H), 2.24 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 166.3, 165.2, 165.1, 138.4, 133.6, 133.5, 133.4, 133.2, 130.0, 129.97, 129.96, 129.8, 129.7, 129.4, 128.7, 128.6, 128.50, 128.48, 86.6, 83.5, 76.3, 71.9, 70.6, 63.5, 60.1, 21.2; HRMS (ESI): m/z calcd for C35H36O8NS [M + NH4]+ 630.2156, found 630.2149.4-Methoxyphenyl 3-O-acetyl-2,4-di-O-benzoyl-β-D-xylopyranoside (31)
Following the procedure for the preparation of compound 27, 30 (1.35 mg, 5.27 mmol) was converted into 31 (1.6 mg, 3.16 mmol, 60%) over two steps by purification with silica gel column (petroleum ether/EtOAc = 6/1). [α]24D = −34.2 (c 0.90, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.03 (d, J = 7.7 Hz, 4H), 7.63–7.51 (m, 2H), 7.48–7.34 (m, 4H), 6.96 (d, J = 9.0 Hz, 2H), 6.80 (d, J = 9.0 Hz, 2H), 5.60 (t, J = 7.6 Hz, 1H), 5.47 (dd, J = 7.6, 5.8 Hz, 1H), 5.31–5.20 (m, 2H), 4.45 (dd, J = 12.2, 4.4 Hz, 1H), 3.75 (s, 3H), 3.70 (dd, J = 12.2, 7.3 Hz, 1H), 2.02 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 170.0, 165.6, 165.3, 155.7, 150.8, 133.7, 133.6, 130.04, 130.02, 129.31, 129.29, 128.7, 128.6, 118.8, 114.7, 99.9, 70.5, 70.3, 69.3, 61.9, 55.8, 20.9; HRMS (ESI): m/z calcd for C28H26O9Na [M + Na]+ 529.1469, found 529.1458.4-Methoxyphenyl 2,4-di-O-benzoyl-β-D-xylopyranoside (7b)
Following the procedure for the preparation of 7a, 7b (85 mg, 0.18 mmol, 93%), purified by silica gel column chromatography (CH2Cl2/EtOAc = 10/1), was prepared from compound 31 (100 mg, 0.20 mmol). [α]24D = −49.0 (c 1.00, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.04 (d, J = 8.0 Hz, 2H), δ 8.03 (d, J = 8.0 Hz, 2H), 7.62–7.50 (m, 2H), 7.44–7.30 (m, 4H), 7.02 (d, J = 8.9 Hz, 2H), 6.83 (d, J = 8.9 Hz, 2H), 5.41 (d, J = 4.1 Hz, 1H), 5.34 (t, J = 4.9 Hz, 1H), 5.24–5.15 (m, 1H), 4.45 (dd, J = 12.8, 3.4 Hz, 1H), 4.25 (t, J = 5.6 Hz, 1H), 3.81 (dd, J = 12.8, 5.0 Hz, 1H), 3.77 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 166.2, 166.0, 155.8, 150.4, 133.6, 133.5, 130.1, 130.0, 129.7, 129.4, 128.5, 118.6, 114.8, 99.0, 71.5, 71.1, 69.4, 60.4, 55.8; HRMS (ESI): m/z calcd for C26H24O8Na [M + Na]+ 487.1363, found 487.1355.4,6-Di-O-benzoyl-3-O-methyl-α-D-glucopyranose-1,2-diyl (4-methoxyphenyl 2,4-di-benzoyl β-D-xylopyranoside)-3-yl orthobenzoate (32)
Following the procedure for the preparation of compound 29, the coupling of 7b (38 mg, 0.083 mmol) and 8 (70 mg, 0.108 mmol) in CH2Cl2 in the presence of TMSOTf (1.5 μL, 0.0083 mmol) furnished 32 (28 mg, 0.029 mmol, 35%) after purification by silica gel column chromatography (toluene/EtOAc = 20/1). [α]24D = −5.3 (c 1.00, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.99 (d, J = 7.6 Hz, 2H), 7.94 (d, J = 7.6 Hz, 2H), 7.85 (d, J = 6.9 Hz, 2H), 7.83 (d, J = 6.9 Hz, 2H), 7.69 (d, J = 7.4 Hz, 2H), 7.58 (t, J = 7.5 Hz, 1H), 7.54 (t, J = 7.5 Hz, 1H), 7.48 (t, J = 7.4 Hz, 1H), 7.42 (t, J = 7.5 Hz, 3H), 7.38–7.28 (m, 7H), 7.17 (t, J = 7.6 Hz, 2H), 6.95 (d, J = 9.0 Hz, 2H), 6.78 (d, J = 9.0 Hz, 2H), 6.04 (d, J = 5.2 Hz, 1H), 5.37 (t, J = 4.6 Hz, 1H), 5.28 (m, 1H), 5.21–5.15 (m, 1H), 5.12 (d, J = 8.9 Hz, 1H), 4.91–4.84 (m, 1H), 4.39 (dd, J = 12.4, 3.3 Hz, 1H), 4.28–4.17 (m, 2H), 4.11 (t, J = 5.3 Hz, 1H), 3.86–3.78 (m, 1H), 3.75 (s, 3H), 3.68 (dd, J = 12.5, 5.2 Hz, 1H), 3.59 (s, 3H), 3.55 (s, 1H); 13C NMR (126 MHz, CDCl3) δ 166.1, 165.5, 165.4, 165.2, 155.4, 150.8, 135.5, 133.5, 133.4, 133.3, 133.0, 130.0, 129.90, 129.87, 129.81, 129.78, 129.76, 129.51, 129.49, 128.5, 128.4, 128.34, 128.29, 126.7, 122.0, 118.4, 114.7, 98.7, 97.9, 77.1, 73.3, 70.8, 70.5, 69.4, 68.5, 67.4, 64.5, 60.6, 58.6, 55.8; HRMS (ESI): m/z calcd for C54H52O16N [M + NH4]+ 970.3281, found 970.3283.4-Methoxyphenyl 2,4,6-tri-O-benzoyl-3-O-methyl-α-D-glucopyranosyl-(1→3)-2,4-di-O-benzoyl-β-D-xylopyranoside (33α) and 4-methoxyphenyl 2,4,6-tri-O-benzoyl-3-O-methyl-β-D-glucopyranosyl-(1→3)-2,4-di-O-benzoyl-β-D-xylopyranoside (33β)
According to the protocol for the synthesis of 29, TBSOTf (6.0 μL, 0.025 mmol)-catalyzed glycosylation of 7b (38 mg, 0.083 mmol) and 8 (70 mg, 0.108 mmol) in anhydrous CH2Cl2 (2 mL) to afford 33α (18 mg, 0.019 mmol, 23%) and 33β (31 mg, 0.032 mmol, 39%) with the purification of silica gel column chromatography (toluene/EtOAc = 30/1) 33α: [α]24D = +66.8 (c 1.00, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.04 (d, J = 7.3 Hz, 2H), 7.99 (d, J = 7.2 Hz, 2H), 7.95 (d, J = 7.3 Hz, 2H), 7.89 (d, J = 7.2 Hz, 2H), 7.79 (d, J = 7.3 Hz, 2H), 7.63–7.42 (m, 7H), 7.39 (s, 2H), 7.35–7.27 (m, 6H), 7.00 (d, J = 9.1 Hz, 2H), 6.81 (d, J = 9.1 Hz, 2H), 5.64 (d, J = 3.7 Hz, 1H), 5.60–5.54 (m, 1H), 5.46 (t, J = 9.7 Hz, 1H), 5.27 (d, J = 4.9 Hz, 1H), 5.19–5.04 (m, 2H), 4.47–4.28 (m, 4H), 4.07 (t, J = 9.7 Hz, 1H), 3.91 (dd, J = 12.2, 4.0 Hz, 1H), 3.77 (s, 3H), 3.56 (dd, J = 12.3, 6.2 Hz, 1H), 3.44 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 166.2, 165.4, 165.2, 165.0, 155.6, 150.8, 133.8, 133.52, 133.49, 133.1, 129.98, 129.93, 129.86, 129.85, 129.5, 129.22, 129.21, 129.0, 128.7, 128.6, 128.53, 128.46, 128.4, 118.5, 114.8, 99.3, 96.9, 78.8, 73.9, 72.8, 70.7, 70.6, 70.5, 68.7, 62.5, 60.9, 60.8, 55.8; HRMS (ESI): m/z calcd for C54H52O16N [M + NH4]+ 970.3281, found 970.3261. 33β: [α]24D = −35.6 (c 0.75, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.04 (d, J = 7.6 Hz, 2H), 8.00 (d, J = 7.5 Hz, 2H), 7.95–7.89 (m, 6H), 7.60–7.40 (m, 6H), 7.36–7.22 (m, 7H), 7.12 (t, J = 7.7 Hz, 2H), 6.73 (d, J = 9.1 Hz, 2H), 6.67 (d, J = 9.1 Hz, 2H), 5.52–5.42 (m, 2H), 5.32 (s, 1H), 5.24 (s, 1H), 5.19–5.12 (m, 2H), 4.57 (dd, J = 12.0, 2.9 Hz, 1H), 4.50–4.37 (m, 3H), 4.15–4.08 (m, 1H), 3.89 (t, J = 9.2 Hz, 1H), 3.78–3.71 (m, 4H), 3.39 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 166.3, 165.5, 165.3, 165.2, 165.1, 155.1, 150.3, 133.6, 133.5, 133.2, 133.1, 132.9, 130.00, 129.97, 129.81, 129.78, 129.72, 129.4, 129.3, 128.7, 128.5, 128.4, 128.33, 128.27, 118.5, 114.4, 110.1, 101.2, 97.2, 82.0, 73.0, 72.69, 72.68, 70.7, 69.1, 68.7, 63.6, 59.6, 58.9, 55.8; HRMS (ESI): m/z calcd for C54H52O16N [M + NH4]+ 970.3281, found 970.3288.Benzyl 2,4,6-tri-O-benzoyl-3-O-methyl-β-D-glucopyranosyl-(1→3)-2,4-di-O-benzoyl-α-D-xylopyranoside (34)
Following the procedure for the preparation of compound 29, the coupling of 7c (1.38 g, 3.07 mmol) and 8 (3.0 g, 4.61 mmol) in anhydrous CH2Cl2 (20 mL) in the presence of TMSOTf (56 μL, 0.022 mmol) afford 34 (2.30 g, 2.46 mmol, 80%) by silica gel column chromatography (toluene/EtOAc = 30/1). [α]24D = +1.0 (c 1.00, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.06 (d, J = 7.2 Hz, 2H), 8.02–7.95 (m, 4H), 7.93 (d, J = 7.2 Hz, 2H), 7.69 (t, J = 7.4 Hz, 1H), 7.63–7.50 (m, 5H), 7.49–7.35 (m, 6H), 7.33–7.22 (m, 3H), 7.20–7.12 (m, 5H), 7.12–7.04 (m, 2H), 5.30 (t, J = 9.5 Hz, 1H), 5.27–5.19 (m, 2H), 5.11 (d, J = 3.5 Hz, 1H), 5.06 (d, J = 8.0 Hz, 1H), 4.96 (dd, J = 9.4, 3.6 Hz, 1H), 4.69 (d, J = 12.3 Hz, 1H), 4.60 (t, J = 9.1 Hz, 1H), 4.48–4.39 (m, 2H), 4.31 (dd, J = 11.9, 5.9 Hz, 1H), 4.08–3.98 (m, 2H), 3.78 (t, J = 10.6 Hz, 1H), 3.70 (t, J = 9.3 Hz, 1H), 3.24 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 166.4, 165.6, 165.2, 165.1, 164.9, 137.0, 133.6, 133.3, 133.2, 133.0, 130.0, 129.93, 129.88, 129.79, 129.77, 129.68, 129.62, 129.59, 129.40, 129.35, 128.6, 128.5, 128.38, 128.35, 128.34, 128.0, 127.8, 101.5, 95.1, 81.9, 75.5, 73.9, 72.7, 72.2, 70.8, 69.9, 69.7, 63.9, 59.5, 59.2; HRMS (ESI): m/z calcd for C54H52O15N [M + NH4]+ 954.3331, found 954.3329.2,4,6-Tri-O-benzoyl-3-O-methyl-β-D-glucopyranosyl-(1→3)-2,4-di-O-benzoyl-α-D-xylopyranosyl trichloroacetimidate (35)
To a solution of 34 (2.58 mg, 2.75 mmol) in EtOH (70 mL) was added 10% Pd/C (1 g). The reaction vessel was evacuated and backfilled with hydrogen (1 atm). The mixture was stirred at 50 °C for 24 h, at this stage TLC indicated the reaction was completed. The mixture was filtered through a pad of Celite. The filtrate was concentrated and the residue was purified by silica gel column chromatography (petroleum ether/EtOAc = 2.5/1) to afford the desired hemiacetal as a white foam which was directly used in the next step. To a soln of the above hemiacetal (1.91 g, 2.26 mmol) and Cl3CCN (2.3 mL, 22.6 mmol) in CH2Cl2 (2.5 mL) was added DBU (171 μL, 1.15 mmol) at 0 °C. The mixture was stirred overnight at ambient temperature, and then concentrated in vacuo. The resulting residue was purified by silica gel column chromatography (petroleum ether/EtOAc = 3.5/1) to afford 35 (2.02 g, 2.04 mmol, 74% over two steps). [α]24D = −15.7 (c 1.00, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.47 (s, 1H), 8.11 (d, J = 7.5 Hz, 2H), 8.01 (d, J = 8.5 Hz, 2H), 7.99 (d, J = 8.5 Hz, 2H), 7.93 (d, J = 7.5 Hz, 2H), 7.71–7.54 (m, 4H), 7.54–7.45 (m, 5H), 7.42 (t, J = 7.7 Hz, 2H), 7.39–7.28 (m, 4H), 7.19 (t, J = 7.7 Hz, 2H), 6.49 (d, J = 3.4 Hz, 1H), 5.38–5.30 (m, 1H), 5.30–5.17 (m, 3H), 5.04 (d, J = 8.0 Hz, 1H), 4.60 (t, J = 9.2 Hz, 1H), 4.54 (dd, J = 12.0, 2.9 Hz, 1H), 4.29–4.17 (m, 2H), 4.12–4.04 (m, 1H), 3.87 (t, J = 10.8 Hz, 1H), 3.70 (t, J = 9.3 Hz, 1H), 3.23 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 166.4, 165.5, 165.3, 164.9, 164.8, 160.5, 133.73, 133.65, 133.5, 133.4, 133.1, 129.98, 129.95, 129.7, 129.6, 129.5, 129.30, 129.29, 129.0, 128.7, 128.6, 128.42, 128.38, 101.6, 93.7, 81.8, 77.2, 75.4, 72.6, 72.55, 72.48, 70.7, 68.8, 64.0, 61.7, 59.3; HRMS (ESI): m/z calcd for C49H46O15N2Cl3 [M + NH4]+ 1007.1958, found 1007.1957.p-Tolyl 2,3-di-O-benzoyl-6-deoxy-6-iodo-1-thio-β-D-glucopyranoside (37)
To a solution of 36 (1.13 g, 2.29 mmol), Ph3P (1.20 g, 4.58 mmol) and imidazole (0.62 g, 9.16 mmol) in anhydrous THF (20 mL) was added a solution of I2 (0.76 g, 2.98 mmol) in anhydrous THF (3 mL). After stirring for 1 h, monitoring by TLC indicated that the reaction went to completion. The mixture was filtered and the filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography to give 37 (petroleum ether/EtOAc = 5/1, 1.26 g, 2.08 mmol, 91%). [α]24D = +52.4 (c 1.00, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.98 (d, J = 8.0 Hz, 2H), 7.94 (d, J = 8.0 Hz, 2H), 7.57–7.45 (m, 4H), 7.44–7.32 (m, 4H), 7.13 (d, J = 7.9 Hz, 2H), 5.45–5.31 (m, 2H), 4.89 (d, J = 9.1 Hz, 1H), 3.79–3.65 (m, 2H), 3.47 (dd, J = 10.7, 6.4 Hz, 1H), 3.42–3.34 (m, 1H), 3.21 (s, 1H), 2.35 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 168.0, 165.3, 138.9, 134.4, 133.9, 133.5, 130.2, 130.0, 129.8, 129.4, 128.7, 128.6, 128.5, 127.5, 86.0, 79.0, 78.4, 73.3, 70.0, 21.4, 5.6; HRMS (ESI): m/z calcd for C27H29O6NIS [M + NH4]+ 622.0755, found 622.0743.p-Tolyl 2,3-di-O-benzoyl-6-deoxy-1-thio-β-D-glucopyranoside (9)
To a solution of 19 (1.38 g, 2.28 mmol) in anydrous N,N-dimethylformamide (45 mL) was added 10% Pd/C (700 mg) and NaHCO3 (633 mg, 7.53 mmol). The reaction vessel was evacuated and backfilled with hydrogen (1 atm). The mixture was stirred at 40 °C for 24 h. The solid was filtered on Celite and the filtrate was concentrated. The crude product was purified by silica gel column chromatography (petroleum ether/EtOAc = 5/1) to give 9 (904 mg, 1.89 mmol, 83%). [α]24D = +89.7 (c 1.00, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.02–7.90 (m, 4H), 7.56–7.48 (m, 2H), 7.42–7.33 (m, 6H), 7.11 (d, J = 8.1 Hz, 2H), 5.40 (t, J = 9.6 Hz, 1H), 5.32 (t, J = 9.0 Hz, 1H), 4.83 (d, J = 9.9 Hz, 1H), 3.65–3.49 (m, 2H), 2.93 (d, J = 4.4 Hz, 1H), 2.34 (s, 3H), 1.46 (d, J = 5.8 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 167.9, 165.4, 138.6, 133.7, 133.6, 133.4, 130.1, 129.9, 129.8, 129.5, 129.0, 128.6, 128.50, 128.49, 86.3, 78.8, 74.7, 70.6, 21.3, 18.0; HRMS (ESI): m/z calcd for C27H30O6NS [M + NH4]+ 496.1788, found 496.1786.p-Tolyl 2,4,6-tri-O-benzoyl-3-O-methyl-β-D-glucopyranosyl-(1→3)-2,4-di-O-benzoyl-β-D-xylopyranosyl-(1→4)-2,3-di-O-benzoyl-6-deoxy-1-thio-β-D-glucopyranoside (4)
A solution of 35 (1.24 g, 1.25 mmol) and 9 (400 mg, 0.84 mmol) in anhydrous CH2Cl2 (12 mL) was stirred vigorously in the presence of activated 4 Å molecular sieves (1 g) for 20 min. The mixture was cooled to 0 °C and TMSOTf (15 μL, 0.084 mmol) were added. After 1 h, the reaction mixture was quenched with Et3N and filtered. The filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc = 3/1) to give 4 (756 mg, 70%). [α]24D = +2.6 (c 1.00, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.01 (s, 2H), 7.98 (s, 2H), 7.96 (d, J = 7.7 Hz, 2H), 7.93 (d, J = 7.7 Hz, 2H), 7.89 (d, J = 7.8 Hz, 4H), 7.80 (d, J = 7.7 Hz, 2H), 7.60–7.07 (m, 24H), 5.48 (t, J = 9.2 Hz, 1H), 5.37 (t, J = 9.4 Hz, 1H), 5.32 (t, J = 8.3 Hz, 1H), 5.23 (t, J = 9.7 Hz, 1H), 5.03–4.97 (m, 1H), 4.97–4.88 (m, 2H), 4.74 (d, J = 2.8 Hz, 1H), 4.63 (d, J = 10.1 Hz, 1H), 4.47 (dd, J = 11.9, 3.0 Hz, 1H), 4.33 (dd, J = 12.0, 6.3 Hz, 1H), 4.21 (t, J = 4.5 Hz, 1H), 4.06–3.96 (m, 1H), 3.93 (dd, J = 12.7, 2.4 Hz, 1H), 3.73 (t, J = 9.2 Hz, 1H), 3.42 (t, J = 9.2 Hz, 1H), 3.32 (s, 3H), 3.22–3.15 (m, 1H), 3.11 (dd, J = 12.7, 4.0 Hz, 1H), 2.35 (s, 3H), 1.10 (t, J = 6.0 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 166.2, 165.6, 165.5, 165.3, 165.2, 164.9, 164.7, 138.4, 133.6, 133.44, 133.37, 133.27, 133.26, 133.19, 133.00, 132.95, 130.02, 129.94, 129.91, 129.8, 129.7, 129.6, 129.4, 129.2, 129.0, 128.63, 128.59, 128.5, 128.43, 128.40, 128.32, 128.27, 101.2, 100.0, 86.2, 82.7, 81.7, 75.4, 74.8, 74.0, 72.7, 72.5, 71.5, 70.6, 70.3, 68.8, 63.7, 59.6, 18.0; HRMS (ESI): m/z calcd for C74H70O20NS [M + NH4]+ 1324.4206, found 1324.4198.Lanost-7-en-3β-yl 2,4,6-tri-O-benzoyl-3-O-methyl-β-D-glucopyranosyl-(1→3)-2,4-di-O-benzoyl-β-D-xylopyranosyl-(1→4)-2,3-di-O-benzoyl-6-deoxy-β-D-glucopyranosyl-(1→2)-3-O-benzoyl-4-O-(2-naphthylmethyl)-β-D-xylopyranoside (2)
A solution of 3 (62 mg, 0.076 mmol) and 4 (150 mg, 0.115 mmol) in anhydrous CH2Cl2 (2 mL) was stirred vigorously in the presence of activated 4 Å molecular sieves (200 mg) for 20 min. The mixture was cooled to 0 °C and NIS (34 mg, 0.152 mmol) and TMSOTf (2 μL, 0.008 mmol) were added. After stirring for 2 h, the reaction mixture was quenched with Et3N, and the solid was filtered off. The filtrate was washed with saturated aqueous Na2S2O3 and brine. The organic phase was dried over Na2SO4, filtered and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc = 3.5/1) to give 2 (134 mg, 67%). [α]24D = +10.6 (c 0.75, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.00 (d, J = 7.3 Hz, 2H), 7.94–7.81 (m, 10H), 7.78 (d, J = 7.3 Hz, 2H), 7.74–7.65 (m, 3H), 7.62 (t, J = 7.4 Hz, 1H), 7.60–7.49 (m, 5H), 7.49–7.43 (m, 5H), 7.43–7.35 (m, 8H), 7.32–7.24 (m, 8H), 7.18 (t, J = 7.8 Hz, 2H), 7.14 (dd, J = 8.4, 1.2 Hz, 1H), 5.36–5.23 (m, 4H), 5.21 (s, 1H), 5.16 (t, 8.1 Hz, 1H), 5.03–4.97 (m, 1H), 4.97–4.93 (m, 1H), 4.87 (d, J = 7.8 Hz, 1H), 4.71 (d, J = 7.9 Hz, 1H), 4.64 (d, J = 4.0 Hz, 1H), 4.59–4.46 (m, 3H), 4.42 (dd, J = 12.0, 3.2 Hz, 1H), 4.28 (dd, J = 11.9, 6.3 Hz, 1H), 4.17 (t, J = 5.6 Hz, 1H), 4.04–3.90 (m, 2H), 3.86–3.75 (m, 2H), 3.67 (t, J = 9.2 Hz, 1H), 3.52–3.61 (m, 1H), 3.39–3.31 (m, 2H), 3.29 (s, 3H), 3.25–3.16 (m, 1H), 3.09 (dd, J = 11.7, 3.6 Hz, 1H), 3.05 (dd, J = 12.6, 5.0 Hz, 1H), 2.04–1.88 (m, 4H), 1.82–1.71 (m, 2H), 1.04 (s, 3H), 1.02–0.97 (m, 6H), 0.91–0.83 (m, 15H), 0.64 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 166.2, 165.6, 165.5, 165.3, 165.2, 164.8, 164.7, 145.1, 135.2, 133.6, 133.42, 133.40, 133.2, 133.1, 133.04, 133.00, 132.8, 132.7, 130.1, 130.0, 129.97, 129.93, 129.90, 129.87, 129.8, 129.7, 129.6, 129.4, 129.2, 128.7, 128.6, 128.5, 128.4, 128.3, 128.23, 128.19, 128.0, 127.7, 126.8, 126.1, 126.0, 125.9, 116.8, 103.7, 101.5, 100.5, 100.4, 90.2, 83.1, 81.7, 76.4, 75.2, 74.6, 74.4, 73.7, 72.70, 72.68, 72.4, 72.1, 71.0, 70.9, 70.6, 69.1, 63.7, 62.5, 60.2, 59.3, 52.1, 51.0, 50.7, 47.3, 44.5, 39.7, 39.2, 38.3, 36.7, 36.6, 35.4, 32.32, 32.26, 28.2, 27.8, 26.3, 25.0, 24.3, 23.0, 22.7, 20.2, 19.1, 17.6, 16.4, 16.2, 14.2; HRMS (ESI): m/z calcd for C120H130O26Na [M + Na]+ 2009.8743, found 2009.8736.Lanost-7-en-3β-yl 2,4,6-tri-O-benzoyl-3-O-methyl-β-D-glucopyranosyl-(1→3)-2,4-di-O-benzoyl-β-D-xylopyranosyl-(1→4)-2,3-di-O-benzoyl-6-deoxy-β-D-glucopyranosyl-(1→2)-3-O-benzoyl-β-D-xylopyranoside (38)
To a solution of 2 (100 mg, 0.05 mmol) in CH2Cl2 (2.80 mL) and MeOH (0.80 mL) was added DDQ (34 mg, 0.15 mmol) in three portions at half an hour interval. After 5 h, the reaction was completed and the volatile was removed by evaporation. The residue was taken up in dichloromethane and washed with saturated aqueous NaHCO3 and brine. The collected organic phase was dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc = 2.5/1) to give 38 (81 mg, 88%). [α]24D = −2.5 (c 1.05, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.11–7.97 (m, 6H), 7.96–7.83 (m, 8H), 7.79 (d, J = 7.3 Hz, 2H), 7.66–7.53 (m, 3H), 7.52–7.38 (m, 10H), 7.38–7.19 (m, 9H), 7.15 (t, J = 7.8 Hz, 2H), 5.48 (t, J = 9.4 Hz, 1H), 5.42–5.31 (m, 2H), 5.26–5.20 (m, 1H), 5.18 (s, 1H), 5.03–4.98 (m, 1H), 4.98–4.95 (m, 1H), 4.94 (d, J = 7.8 Hz, 1H), 4.88 (t, J = 4.0 Hz, 1H), 4.82 (s, 1H), 4.76–4.67 (m, 2H), 4.47 (dd, J = 12.0, 3.2 Hz, 1H), 4.34 (dd, J = 12.0, 6.3 Hz, 1H), 4.25 (dd, J = 10.1, 1.8 Hz, 1H), 4.22 (t, J = 4.7 Hz, 1H), 4.04–3.97 (m, 1H), 3.94 (dd, J = 12.6, 2.6 Hz, 1H), 3.90 (s, 1H), 3.75 (t, J = 9.2 Hz, 1H), 3.64–3.56 (m, 1H), 3.53 (dd, J = 12.3, 2.9 Hz, 1H), 3.43 (t, J = 9.2 Hz, 1H), 3.33 (s, 3H), 3.19–3.03 (m, 4H), 1.01 (d, J = 6.0 Hz, 3H), 0.97 (s, 3H), 0.91–0.85 (m, 12H), 0.84 (s, 3H), 0.67 (s, 3H), 0.64 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 166.2, 166.04, 166.00, 165.5, 165.3, 165.2, 164.9, 164.7, 145.2, 133.6, 133.6, 133.5, 133.4, 133.2, 133.2, 133.1, 133.0, 130.1, 130.00, 129.96, 129.9, 129.78, 129.75, 129.74, 129.6, 129.42, 129.38, 129.2, 128.7, 128.6, 128.5, 128.44, 128.43, 128.33, 128.29, 116.6, 102.1, 101.3, 101.0, 100.1, 89.6, 82.9, 81.7, 77.4, 73.9, 73.7, 73.2, 72.72, 72.69, 72.5, 71.2, 70.6, 70.3, 68.8, 67.1, 63.7, 61.7, 59.6, 59.3, 52.1, 51.0, 50.5, 47.3, 44.5, 39.7, 39.1, 38.3, 36.7, 36.6, 35.4, 32.3, 32.2, 28.3, 28.2, 27.8, 26.2, 25.0, 24.3, 23.00, 22.98, 22.7, 20.2, 19.1, 17.5, 16.3, 16.2, 14.3; HRMS (ESI): m/z calcd for C109H126O26N [M + NH4]+ 1864.8563, found 1864.8521.Lanost-7-en-3β-yl 2,4,6-tri-O-benzoyl-3-O-methyl-β-D-glucopyranosyl-(1→3)-2,4-di-O-benzoyl-β-D-xylopyranosyl-(1→4)-2,3-di-O-benzoyl-6-deoxy-β-D-glucopyranosyl-(1→2)-3-O-benzoyl-4-O-sulfo-β-D-xylopyranoside (39)
To a mixture of alcohol 38 (60 mg, 0.032 mmol) in anhydrous pyridine (2.0 mL) was added sulfur trioxide/pyridine complex (103 mg, 0.65 mmol). The sealed Pyrex tube was irradiated in the microwave (100 W, CEM corporation, Matthews, North Carolina, USA) at 100 °C for 2 h. After cooling, water (1 mL) was added to the mixture, which was then concentrated in vacuo. The residue was purified by silica gel column chromatography (CH2Cl2/MeOH = 13/1) to give 39 (60 mg, 0.03 mmol, 94%). [α]24D = +2.8 (c 1.00, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.04 (d, J = 5.4 Hz, 2H), 7.99 (d, J = 7.5 Hz, 2H), 7.97 (d, J = 7.5 Hz, 2H), 7.93 (d, J = 7.6 Hz, 2H), 7.87 (d, J = 6.5 Hz, 4H), 7.79 (d, J = 7.5 Hz, 2H), 7.63 (d, J = 7.6 Hz, 2H), 7.60–7.52 (m, 3H), 7.50 (t, J = 7.3 Hz, 1H), 7.47–7.30 (m, 11H), 7.29–7.19 (m, 4H), 7.15 (t, J = 7.6 Hz, 2H), 7.10–7.00 (m, 1H), 6.73–6.54 (m, 2H), 5.46 (t, J = 9.0 Hz, 1H), 5.39–5.24 (m, 3H), 5.19 (s, 1H), 5.04–4.97 (m, 2H), 4.97–4.81 (m, 3H), 4.72 (s, 1H), 4.61 (d, J = 7.8 Hz, 1H), 4.52–4.39 (m, 2H), 4.34 (d, J = 11.9 Hz, 1H), 4.29 (dd, J = 11.8, 6.1 Hz, 1H), 4.20 (t, J = 4.8, 1H), 3.99–3.88 (m, 2H), 3.88–3.78 (m, 2H), 3.76–3.60 (m, 1H), 3.49 (t, J = 9.0 Hz, 1H), 3.29 (s, 3H), 3.16–3.08 (m, 1H), 3.08–2.98 (m, 2H), 1.00 (d, J = 5.5 Hz, 3H), 0.98 (s, 3H), 0.83 (s, 3H), 0.64 (s, 3H), 0.62 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 166.2, 165.7, 165.4, 165.3, 165.2, 164.9, 164.7, 145.1, 133.5, 133.4, 133.3, 133.2, 133.0, 130.3, 130.03, 129.93, 129.88, 129.71, 129.70, 129.6, 129.5, 129.4, 129.2, 128.9, 128.64, 128.60, 128.5, 128.4, 128.3, 128.0, 127.9, 116.6, 102.16, 102.14, 101.1, 100.5, 90.0, 82.6, 81.7, 75.0, 74.4, 73.7, 72.8, 72.7, 72.4, 71.4, 71.2, 70.7, 70.6, 68.8, 63.7, 62.9, 60.0, 59.3, 52.1, 51.0, 50.5, 47.3, 44.5, 39.7, 39.1, 38.3, 36.7, 36.6, 35.4, 32.3, 32.2, 32.1, 30.3, 29.8, 29.7, 29.5, 28.5, 28.1, 27.8, 26.1, 25.0, 24.3, 23.0, 22.8, 22.7, 20.2, 19.1, 17.5, 16.2, 16.2, 14.2; HRMS (ESI): m/z calcd for C109H122O29S [M − Na]− 1926.7787, found 1926.7743.Sodium salt of lanost-7-en-3β-yl 3-O-methyl-β-D-glucopyranosyl-(1→3)-β-D-xylopyranosyl-(1→4)-6-deoxy-β-D-glucopyranosyl-(1→2)-4-O-sulfo-β-D-xylopyranoside (1)
To a solution 39 (50 mg, 0.026 mmol) in 9 mL of anhydrous methanol/CH2Cl2 (v/v = 2/1) was added a solution of sodium methoxide (1.04 mL, 1.04 mmol, 1 M) in methanol. After the resulting mixture was stirred at rt for 30 h, it was diluted with water, and then concentrated under reduced pressure. The residue was dialyzed against water followed by lyophilization to afford 1 (25 mg, 86%). [α]24D = −7.0 (c 0.25, H2O); 1H NMR (500 MHz, pyridine-d5) δ 5.40 (d, J = 7.6 Hz, 1H), 5.35 (s, 1H), 5.26–4.90 (m, 4H), 4.87 (d, J = 7.3 Hz, 1H), 4.81–4.63 (m, 2H), 4.49 (d, J = 10.6 Hz, 1H), 4.40–4.19 (m, 4H), 4.18–3.93 (m, 7H), 3.88 (s, 3H), 3.82–3.53 (m, 5H), 3.26 (d, 1H), 1.74 (d, J = 5.4 Hz, 3H), 1.26 (s, 3H), 1.19 (s, 3H), 1.11 (s, 3H), 0.99 (d, J = 8.6 Hz, 3H), 0.98 (s, 3H), 0.92 (d, J = 6.5 Hz, 6H), 0.76 (s, 3H); 13C NMR (126 MHz, pyridine-d5) δ 145.8, 117.7, 106.0, 105.9, 105.7, 89.6, 88.5, 87.6, 86.5, 84.0, 78.7, 76.8, 76.2, 76.1, 75.8, 75.6, 74.3, 72.3, 71.1, 69.4, 67.1, 64.9, 62.5, 61.4, 52.8, 51.7, 51.4, 48.0, 45.2, 40.3, 40.0, 38.8, 37.33, 37.27, 36.0, 33.1, 32.9, 28.8, 28.7, 28.4, 27.5, 25.5, 25.0, 23.7, 23.5, 23.2, 20.8, 19.7, 18.5, 17.1, 16.8, 14.9; HRMS (ESI): m/z calcd for C53H89O21S [M − Na]− 1093.5621, found 1093.5625.Acknowledgements
We are grateful for the financial support from NSFC-Shandong Joint Fund (U1406402); National Science Funding of China (21272220); Project on Scientific development in Shandong Province (2012GHY11526) and Fundamental Research Funds for the Central Universities. We also thank Dr Patrick Chaffey and Dr Zhongping Tan at the University of Colorado Boulder for their revisions of this manuscript.Notes and references
- K. Hostettmann and A. Marston, Saponins, Cambridge University Press, New York, 1995 Search PubMed.
- (a) V. I. Kalinin, A. S. Silchenko, S. A. Avilov, V. A. Stonik and A. V. Smirnov, Phys. Rev., 2005, 4, 221–236 CAS; (b) S.-K. Kim, S. W. A. Himaya and K.-H. Kang, Marine Pharmacognosy Trends and Applications, ed. S.-K. Kim, CRC Press, Boca Raton, 2013, pp. 119–127 Search PubMed; (c) D. L. Aminin, E. A. Pislyagin, E. S. Menchinskaya, A. S. Silchenko, S. A. Avilov and V. I. Kalinin, Studies in Natural Product Chemistry, ed. A. Rahman, Elsevier, Kidlington, 2014, pp. 75–94 Search PubMed.
- (a) Y.-H. Yi, Q.-Z. Xu, L. Li, S.-L. Zhang, H.-M. Wu, J. Ding, Y.-G. Tong, W.-F. Tan, M.-H. Li, F. Tian, J.-H. Wu, C.-C. Liaw, K. F. Bastow and K.-H. Lee, Helv. Chim. Acta, 2006, 89, 54–63 CrossRef; (b) S.-L. Zhang, L. Li, Y.-H. Yi and P. Sun, Nat. Prod. Res., 2006, 20, 399–407 CrossRef CAS PubMed.
- F. Tian, C.-H. Zhu, X.-W. Zhang, X. Xie, X.-L. Xin, Y.-H. Yi, L.-P. Lin, M.-Y. Geng and J. Ding, Mol. Pharmacol., 2007, 72, 545–552 CrossRef CAS PubMed.
- (a) H. Pellissier, Tetrahedron, 2004, 60, 5123–5162 CrossRef CAS; (b) B. Yu, Y. Zhang and P. Tang, Eur. J. Org. Chem., 2007, 5145–5161 CrossRef CAS; (c) B. Yu and J. Sun, Chem.–Asian J., 2009, 4, 642–654 CrossRef CAS PubMed; (d) C. Gauthier, J. Legault and A. Pichette, Org. Chem., 2009, 6, 321–344 CAS; (e) C. Gauthier, J. Legault, M. Piochon-Gauthier and A. Pichette, Phytochem. Rev., 2011, 10, 521–544 CrossRef CAS; (f) B. Yu, J. Song and X. Yang, Acc. Chem. Res., 2012, 45, 1227–1236 CrossRef CAS PubMed; (g) Y. Yang, S. Laval and B. Yu, Adv. Carbohydr. Chem. Biochem., 2014, 71, 137–226 CrossRef PubMed; (h) L. Zu, Y. Zhao and G. Gu, J. Carbohydr. Chem., 2014, 33, 269–297 CrossRef CAS; (i) Y. Yang, X. Zhang and B. Yu, Nat. Prod. Rep., 2015, 32, 1331–1355 RSC.
- For the synthesis of lanostano-18,20-lactone aglycones, see: (a) G. Habermehl, K.-H. Seib and K.-P. Swiderskjj, Liebigs Ann. Chem., 1978, 419–426 CrossRef CAS; (b) G. G. Habermehl and J. H. Kirsch, Toxicon, 1983, 179–181 CrossRef CAS. For the recent efforts to synthesis of sea cucumber triterpene glycosides, see: (c) J. Yu and B. Yu, Chin. Chem. Lett., 2015, 26, 1331–1335 CrossRef CAS.
- A. V. Demchenko, Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance, ed. A. V. Demchenko, WILEY-VCH, Weinheim, 2008, pp. 1–28 Search PubMed.
- (a) Y.-P. Hu, Y.-Q. Zhong, Z.-G. Chen, C.-Y. Chen, Z. Shi, M. M. L. Zulueta, C.-C. Ku, P.-Y. Lee, C.-C. Wang and S.-C. Hung, J. Am. Chem. Soc., 2012, 134, 20722–20727 CrossRef CAS PubMed; (b) G. Xiao and B. Yu, Chem.–Eur. J., 2013, 19, 7708–7712 CrossRef CAS PubMed; (c) S. U. Hansen, G. J. Miller, G. C. Jayson and J. M. Gardiner, Org. Lett., 2013, 15, 88–91 CrossRef CAS PubMed; (d) Y. Dai and B. Yu, Chem. Commun., 2015, 51, 13826–13829 RSC.
- (a) M. Masaoud, J. Schmidt and G. Adam, Phytochemistry, 1995, 38, 795–796 CrossRef CAS; (b) C. Areche, P. Cejas, P. Thomas, A. San-Martín, L. Astudillo, M. Gutiérrez and L. A. Loyola, Quim. Nova, 2009, 32, 2023–2025 CrossRef CAS.
- (a) R. E. Marker, E. L. Wittle and L. W. Mixon, J. Am. Chem. Soc., 1937, 59, 1368–1373 CrossRef CAS; (b) R. B. Woodward, A. A. Patchett, D. H. R. Barton, D. A. J. Ives and R. B. Kelly, J. Chem. Soc., 1957, 1131–1144 RSC.
- B. B. Shingate, B. G. Hazra, D. B. Salunke, V. S. Pore, F. Shirazi and M. V. Deshpande, Tetrahedron, 2013, 69, 11155–11163 CrossRef CAS.
- V. A. Ignatenko and G. P. Tochtrop, J. Org. Chem., 2013, 78, 3821–3831 CrossRef CAS PubMed.
- (a) K. Takahashi, K. Usami, T. Takahashi, T. Okada and M. Morisaki, Chem. Pharm. Bull., 1987, 35, 3467–3469 CrossRef CAS PubMed; (b) G. G. Habermehl, J. H. Kirsch and K. J. Reibstein, Heterocycles, 1982, 17, 183–189 CrossRef CAS.
- CCDC 1419939 (14·MeOH)†.
- M. Balci, Basic 1H- and 13C-NMR Spectroscopy, Elsevier, Kidlington, 2005, pp. 87–133 Search PubMed.
- (a) D. H. R. Barton and S. W. McCombie, J. Chem. Soc., Perkin Trans. 1, 1975, 1574–1585 RSC; (b) N. Martin and E. J. Thomas, Org. Biomol. Chem., 2012, 10, 7952–7964 RSC; (c) M. M. Heravi, A. Bakhtiari and Z. Faghihi, Curr. Org. Synth., 2014, 11, 787–823 CrossRef CAS.
- M. S. Reddy, H. Zhang, S. Phoenix and P. Deslongchamps, Chem.–Asian J., 2009, 4, 725–741 CrossRef CAS PubMed.
- Q. Liu, T. Guo, L. Zhang, Y. Yu, P. Wang, J. Yang and Y. Li, Eur. J. Med. Chem., 2013, 63, 511–522 CrossRef PubMed.
- R. H. Purdy, A. L. Morrow, J. R. Blinn and S. M. Paul, J. Med. Chem., 1990, 33, 1572–1581 CrossRef PubMed.
- (a) P. J. Stang, M. Hanack and L. R. Subramanian, Synthesis, 1982, 85–126 CrossRef CAS; (b) E. R. Binkley and R. W. Binkley, Preparative Carbohydrate Chemistry, ed. S. Hanessian, Marcel Dekker Inc., New York, 1997, pp. 87–104 Search PubMed.
- R. R. Kumar, S. D. Haveli and H. B. Kagan, Synlett, 2011, 1709–1712 Search PubMed.
- T. Itoh, T. Tamura and T. Matsumoto, Steroids, 1976, 27, 275–285 CrossRef CAS PubMed.
- B. Yang, K. Yoshida, Z. Yin, H. Dai, H. Kavunja, M. H. Eldakdouki, S. Sungsuwan, S. B. Dulaney and X. Huang, Angew. Chem., Int. Ed., 2012, 51, 10185–10189 CrossRef CAS PubMed.
- (a) S. David and S. Hanessian, Tetrahedron, 1985, 41, 643–663 CrossRef CAS; (b) R. F. Helm, J. Ralph and L. Anderson, J. Org. Chem., 1991, 56, 7015–7021 CrossRef CAS.
- (a) K. R. Love, R. B. Andrade and P. H. Seeberger, J. Org. Chem., 2001, 66, 8165–8176 CrossRef CAS PubMed; (b) W. Peng, J. Sun, X. Han and B. Yu, Synlett, 2004, 259–262 CAS; (c) K. R. Love and P. H. Seeberger, J. Org. Chem., 2005, 70, 3168–3177 CrossRef CAS PubMed.
- B. Ren, M. Rahm, X. Zhang, Y. Zhou and H. Dong, J. Org. Chem., 2014, 79, 8134–8142 CrossRef CAS PubMed.
- I. Lundt, C. Pedersen and B. Tronier, Acta Chem. Scand., 1964, 18, 1917–1922 CrossRef.
- (a) H. Yu, B. Yu, X. Wu, Y. Hui and X. Han, J. Chem. Soc., Perkin Trans. 1, 2000, 1445–1453 RSC; (b) T. Zhu and G.-J. Boons, Carbohydr. Res., 2000, 329, 709–715 CrossRef CAS PubMed; (c) Z. Li and J. C. Gildersleeve, J. Am. Chem. Soc., 2006, 128, 11612–11619 CrossRef CAS PubMed; (d) S.-S. Chang, C.-H. Shih, K.-C. Lai and K.-K. T. Mong, Chem.–Asian J., 2010, 5, 1152–1162 CrossRef CAS PubMed.
- J. Tamura, A. Yamaguchi, J. Tanaka and Y. Nishimura, J. Carbohydr. Chem., 2007, 26, 61–82 CrossRef CAS.
- F. Kong, Carbohydr. Res., 2007, 342, 345–373 CrossRef CAS PubMed.
- (a) E. Petráková and J. Schraml, Chem. Commun., 1983, 48, 877–888 Search PubMed; (b) A. D. Bruyn, M. Anteunis, R. V. Rijsbergen, M. Claeyssens and P. Kováč, J. Carbohydr. Chem., 1983, 1, 301–309 CrossRef; (c) R. F. Helm and J. Ralph, Carbohydr. Res., 1993, 240, 23–38 CrossRef CAS PubMed; (d) H. Yuasa, N. Miyagawa, M. Nakatani, M. Izumi and H. Hashimoto, Org. Biomol. Chem., 2004, 2, 3548–3556 RSC.
- (a) S. Karamat and W. M. F. Fabian, J. Phys. Chem. A, 2006, 110, 7477–7484 CrossRef CAS PubMed; (b) Ž. J. Vitnik and W. M. F. Fabian, Comput. Theor. Chem., 2015, 1051, 104–109 CrossRef.
- (a) B. Yu and Z. Yang, Org. Lett., 2001, 3, 377–379 CrossRef CAS PubMed; (b) Q. Yang, M. Lei, Q.-J. Yin and J.-S. Yang, Carbohydr. Res., 2007, 342, 1175–1181 CrossRef CAS PubMed.
- M.-C. Yan, Y.-N. Chen, H.-T. Wu, C.-C. Lin, C.-T. Chen and C.-C. Lin, J. Org. Chem., 2007, 72, 299–302 CrossRef CAS PubMed.
- (a) A. Raghuraman, M. Riaz, M. Hindle and U. R. Desai, Tetrahedron Lett., 2007, 48, 6754–6758 CrossRef CAS PubMed; (b) R. A. Al-Horani, P. Ponnusamy, A. Y. Mehta, D. Gailani and U. R. Desai, J. Med. Chem., 2013, 56, 867–878 CrossRef CAS PubMed; (c) P. Xu, W. Xu, Y. Dai, Y. Yang and B. Yu, Org. Chem. Front., 2014, 1, 405–414 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1419939. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra25845f |
| This journal is © The Royal Society of Chemistry 2016 |