
Synthesis of multisubstituted furans via Cu(I)-catalyzed annulation of ketones with alkynoate under ligand- and additive-free conditions†
Zaigang Luo*a,
Yuyu Fanga,
Yu Zhaoa,
Peng Liub,
Xuemei Xua,
Chengtao Fenga,
Zhong Lia and
Jie He*a
aCollege of Chemical Engineering, AnHui University of Science & Technology, Huainan 232001, P. R. China. E-mail: luozi139@163.com; aust_jhe@163.com
bGuang Zhou Institutes of Biomedicine and Health, Chinese Academy of Sciences, Guangzhou 510530, China
First published on 8th January 2016
Abstract
A facile and efficient annulation strategy for the synthesis of multisubstituted furan derivatives has been achieved under mild conditions. The developed transformation via C(sp3)–H bond functionalization catalyzed by copper(I) salts using benzoyl peroxide as an external oxidant possess some obvious advantages. This ligand- and additive-free cyclization protocol offers an environmentally friendly and efficient access to biologically important scaffolds from readily available substrates.
Introduction
Multisubstituted furans represent an important class of five membered heterocycles ubiquitous in natural products, pharmaceuticals, and agrochemicals as well as act as useful intermediates in organic synthesis.1 Hence, the development of efficient methods for their preparation has been an important research orientation in organic chemistry. The classical approaches such as Paal–Knorr synthesis2 and Feist–Benary synthesis3 have already been established for the assembly of multisubstituted furans. The Paal–Knorr method relies on acid catalyzed intramolecular cyclization of 1,4-dicarbonyl compounds and Feist–Benary method represents one of first approaches for the intermolecular annulation of 1,3-dicarbonyl compounds and α-halogen ketones.In the past few decades, transition-metal-catalyzed approaches to the synthesis of furan derivatives have been developed, such as Pd-catalyzed cyclization of 2,3-allenoic acids in the presence of allenes,4 Pd-catalyzed cyclization of alkynylbenziodoxolones and imine derivatives of acetophenone,5 Au-catalyzed cycloisomerizations of bromoallenyl ketones,6 Co-catalyzed cyclization of alkynes and α-diazocarbonyls,7 Ag-catalyzed cycloisomerizations of phenyl and tert-butyl alkynyl ketones utilizing [3,3] acyloxy migration,8 successive Ru-catalyzed dimerization of terminal alkynes and Cu(II)-catalyzed cyclization of 1,3-dienyl ethers9 and so forth. However, the synthetic potential of these strategies is still suffer from limitations like the requirement of functionalized precursors, harsh conditions and low conversions. As a consequence, the development of a new synthetic route to synthesize furan derivatives from simple and commercially available starting materials is still desirable.
Despite the recent advances with these transition metal catalysts, copper catalyzed synthesis of furans is highly attractive. Because copper salts are inexpensive and possess low toxicity. Recently, copper catalyzed C–H functionalization reactions to the construction of heterocycles have been successfully developed.10 In 2010, Jiang' group demonstrated the synthesis of furans from alkynoates and 1,3-dicarbonyl compounds through Sn(II)- and Cu(I)-involved addition/oxidative cyclization (Scheme 1a).11 Subsequently, they reported a novel and convenient one-pot Cu(I)-catalyzed approach for the preparation of 2-carbonyl furans via (2-furyl)carbene complexes.12 Very recently, Antonchick and Manna described the first copper-catalyzed annulation of acetophenone derivatives and an alkyl acetylenedicarboxylate with a broad reaction scope (Scheme 1b).13 This reaction provides a useful method for the synthesis of multisubstituted furans from readily available acetophenones and electron-deficient alkynes via Cu(I)-catalyzed direct C(sp3)–H bond functionalization under radical reaction conditions, which are generally difficult to access from other conventional methods. However, this reaction was proceeded in the presence of a ligand such as 2,2′-bipyridine, and the ligand was essential for the catalytic activity of the copper(I) salt. Moreover, the substrate scope is limited to the electron-poor arylmethyl ketones. Hence, copper catalyzed C(sp3)–H functionalization reactions between arylmethyl ketones and alkynoate to the construction of furans is still filled with challenges.
 | ||
| Scheme 1 Synthesis of furan from ketone and alkynoate. | ||
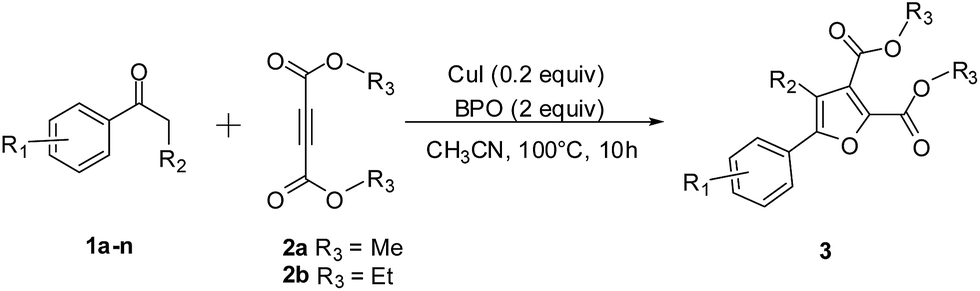
Herein we present a facile and efficient Cu(I)-catalyzed annulation of simple and readily available ketones with alkynoate under mild conditions (Scheme 1c). Comparing to the research work of Antonchick and Manna's,14 the advantages of our current study are clear, such as ligand- and additive-free, high yield, large-scale processes and broad substrate scope (well suitable for both nonactivated arylmethyl ketones with electron-donating or electron-withdrawing groups and 1,3-dicarbonyl compounds with an activated methylene group).
Results and discussion
Initially, acetophenone 1a and dimethyl acetylenedicarboxylate (DMAD) 2a were chosen as the model substrates to optimize the reaction conditions, including the catalyst, oxidant, solvent, and temperature under ambient air. As described in Table 1, six oxidants, air, O2, H2O2, tert-butyl hydroperoxide (TBHP), di-tert-butyl peroxide (DTBP) and benzoyl peroxide (BPO) were investigated at 100 °C in 2 mL of CH3CN using CuI as the catalyst, and BPO was found to be the most effective oxidant (90%) (entries 1–6). When the reaction was carried out without CuI, a significant decrease of the yield was observed (entry 7). Changing the counterion of copper salt, such as CuCl and CuBr (entries 8–9), or replacing Cu(I) with Cu(II), such as CuI2, CuCl2·2H2O, CuBr2, CuCO3, led to inferior results (entries 10–13). Variations in the loading of CuI and BPO did not provide an improvement in yield of product 3aa (entries 14–17). Afterward, the solvents, including CH3CN, DMF, CH3CN/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), DMSO, CH3OH, 1,2-dichloroethane (DCE) and N,N-dimethylacetamide (DMA) were tested using CuI as the catalyst and BPO as the oxidant at 100 °C, and CH3CN provided the highest yield (entries 6, 18–23). Finally, when the temperature was dropped to 90 °C, the target product was not be found (entry 24), and the yield dramatically decreased when the temperature was elevated to 110 °C (entry 25). After the optimization process for catalyst, oxidant, solvent, and temperature, the multisubstituted furans 3 were synthesized under our standard conditions: 0.2 equiv. of CuI as the catalyst, 2 equiv. of BPO as the oxidant, and CH3CN as the solvent under air atmosphere at 100 °C.
:1), DMSO, CH3OH, 1,2-dichloroethane (DCE) and N,N-dimethylacetamide (DMA) were tested using CuI as the catalyst and BPO as the oxidant at 100 °C, and CH3CN provided the highest yield (entries 6, 18–23). Finally, when the temperature was dropped to 90 °C, the target product was not be found (entry 24), and the yield dramatically decreased when the temperature was elevated to 110 °C (entry 25). After the optimization process for catalyst, oxidant, solvent, and temperature, the multisubstituted furans 3 were synthesized under our standard conditions: 0.2 equiv. of CuI as the catalyst, 2 equiv. of BPO as the oxidant, and CH3CN as the solvent under air atmosphere at 100 °C.
|
||||
|---|---|---|---|---|
| Entry | Catalyst (eq.) | Oxidant (eq.) | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.5 mmol), 2a (1 mmol), solvent (2 mL), 100 °C, 10 h.b Isolated yield.c 90 °C.d 110 °C, n.r. – no reaction. | ||||
| 1 | CuI(0.2) | Air | CH3CN | n.r. |
| 2 | CuI(0.2) | O2 | CH3CN | 5 |
| 3 | CuI(0.2) | H2O2(2) | CH3CN | 13 |
| 4 | CuI(0.2) | TBHP(2) | CH3CN | 15 |
| 5 | CuI(0.2) | DTBP(2) | CH3CN | 26 |
| 6 | CuI(0.2) | BPO(2) | CH3CN | 90 |
| 7 | — | BPO(2) | CH3CN | 22 |
| 8 | CuCl(0.2) | BPO(2) | CH3CN | 16 |
| 9 | CuBr(0.2) | BPO(2) | CH3CN | 37 |
| 10 | CuI2(0.2) | BPO(2) | CH3CN | 70 |
| 11 | CuCl2·2H2O (0.2) | BPO(2) | CH3CN | 54 |
| 12 | CuBr2(0.2) | BPO(2) | CH3CN | 56 |
| 13 | CuCO3(0.2) | BPO(2) | CH3CN | 50 |
| 14 | CuI(0.3) | BPO(2) | CH3CN | 85 |
| 15 | CuI(0.1) | BPO(2) | CH3CN | 55 |
| 16 | CuI(0.1) | BPO(1) | CH3CN | 52 |
| 17 | CuI(0.2) | BPO(1) | CH3CN | 83 |
| 18 | CuI(0.2) | BPO(2) | DMF | 54 |
| 19 | CuI(0.2) | BPO(2) | CH3CN:H2O(1:1) |
68 |
| 20 | CuI(0.2) | BPO(2) | DMSO | 52 |
| 21 | CuI(0.2) | BPO(2) | CH3OH | 32 |
| 22 | CuI(0.2) | BPO(2) | DCE | 34 |
| 23 | CuI(0.2) | BPO(2) | DMA | 48 |
| 24c | CuI(0.2) | BPO(2) | CH3CN | n.r. |
| 25d | CuI(0.2) | BPO(2) | CH3CN | 28 |

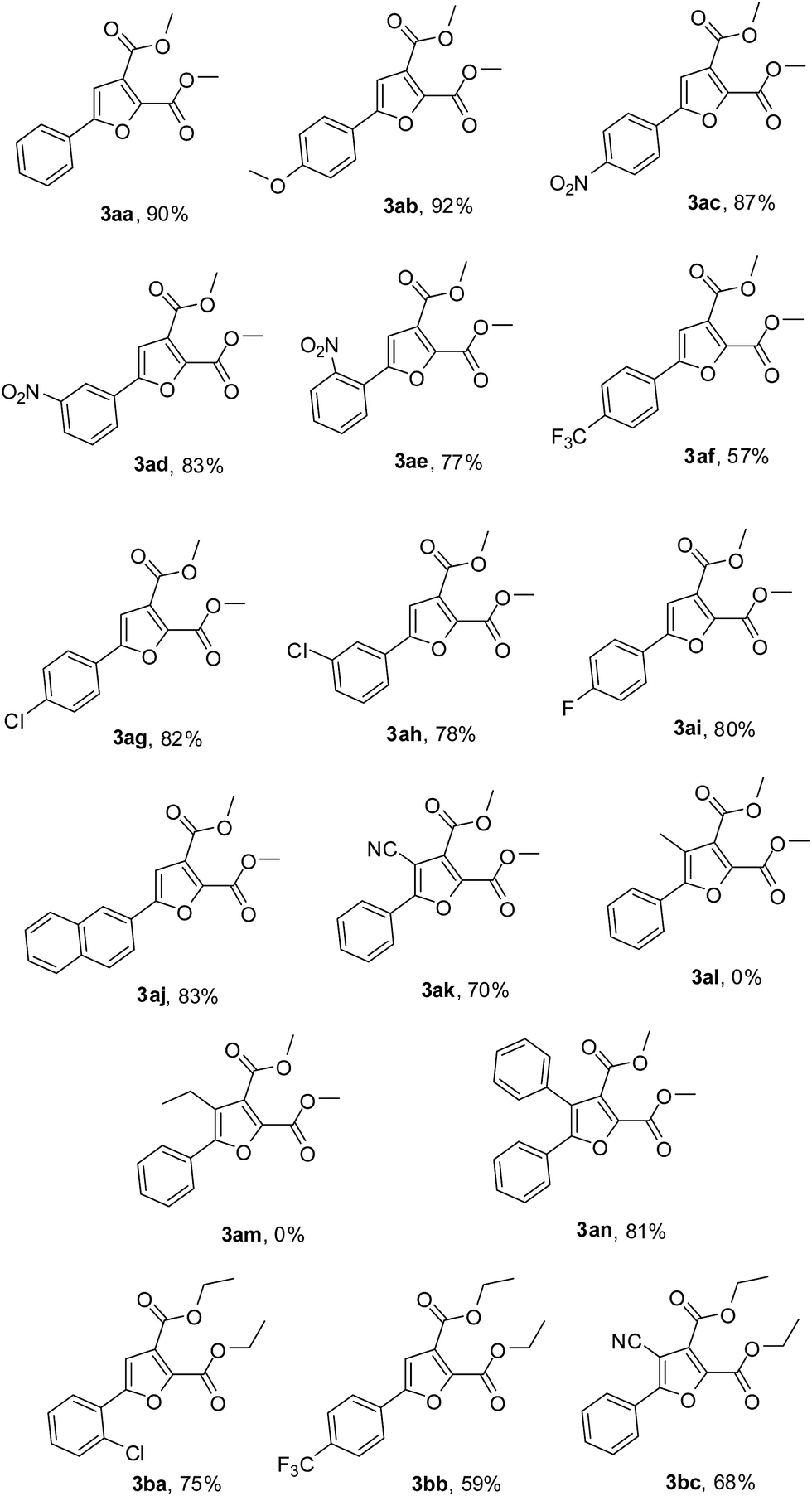
Having established the reaction conditions, various arylmethyl ketones were examined in the cyclization with DMAD (Table 2). To our delight, aryl methyl ketones with electron-donating substituents (p-OMe) or electron-withdrawing substituents (p-NO2, m-NO2, o-NO2, p-CF3), as well as the para-, ortho- and meta-substituted groups both gave the desired products in good yields (3ab–3af), showing no obvious electronic effect in this reaction. While electron-rich aryl methyl ketones were not well tolerated under reported conditions.13 The para- and meta-substituted halogen atoms such as chlorine and fluorine were well tolerated, affording the corresponding products in 82%, 78% and 80% isolated yields, respectively (3ag–3ai). Notably, 2-acetonaphthone 1j is also a suitable substrate for this reaction, which reacted smoothly to give the expected annulation products in good yields (3aj, 83%). Interestingly, benzoylacetonitrile 1k undergoes the annulation and the desired product 3ak was isolated with 70% yield under the reaction conditions. However, when R2 was an electron-donating group, such as methyl or ethyl, the corresponding product 3al and 3am could not be obtained. Obviously, a-position of acetophenones bearing an electron-withdrawing group can afford the desired products smoothly. It is also worth noting that the reaction was insensitive to the sterically hindered substrate 2-phenyl acetophenone 1n and the corresponding product 3an was obtained in 81% isolated yield. In addition, the presence of ethyl groups in acetylenedicarboxylate, diethyl acetylenedicarboxylate 2b, also allowed the synthesis of the corresponding furan derivatives (3ba–3bc). Generally speaking, aryl methyl ketones without sterically hindered groups were cyclized in higher yields than those with sterically hindered groups or sterically hindered substrate.
|
|---|
| a Reaction conditions: arylmethyl ketones 1 (0.5 mmol), 2 (1 mmol), CH3CN (2 mL). Isolated yield. |
 |
Afterward, the application of our present protocol for cyclization of 1,3-dicarbonyl compound and β-ketoester with DMAD, including acetylacetone 1o and ethyl acetoacetate 1p, were explored (Scheme 2). Gratifyingly, the target furan derivatives 3ao and 3ap were isolated in 89% and 85% yields, respectively. In terms of the NMR spectra and related literature,11,15 the structure of 3ap was confirmed, and there was no evidence indicating that the regioisomer of 3ap existed. Obviously, this developed transformation via C(sp3)–H functionalization catalyzed by copper(I) salts using benzoyl peroxide as an external oxidant is well suitable for both nonactivated arylmethyl ketones with electron-donating or electron-withdrawing groups and 1,3-dicarbonyl compounds with an activated methylene group.
 | ||
| Scheme 2 Synthesis of furan from 1,3-dicarbonyl compounds and alkynoate. | ||
Gram-scale applications for the present method were also explored. As shown in Scheme 3, the proposed reaction of 1a and 2a was investigated under the standard conditions, which could give 2.44 g of 3aa in 86% yield without any significant loss of reactive efficiency. Thus, this simple, ligand- and additive-free protocol could be extended as an efficient and practical method to construct various potentially bioactive multisubstituted furan derivatives.
 | ||
| Scheme 3 Synthesis of 3aa in a Gram scale. | ||
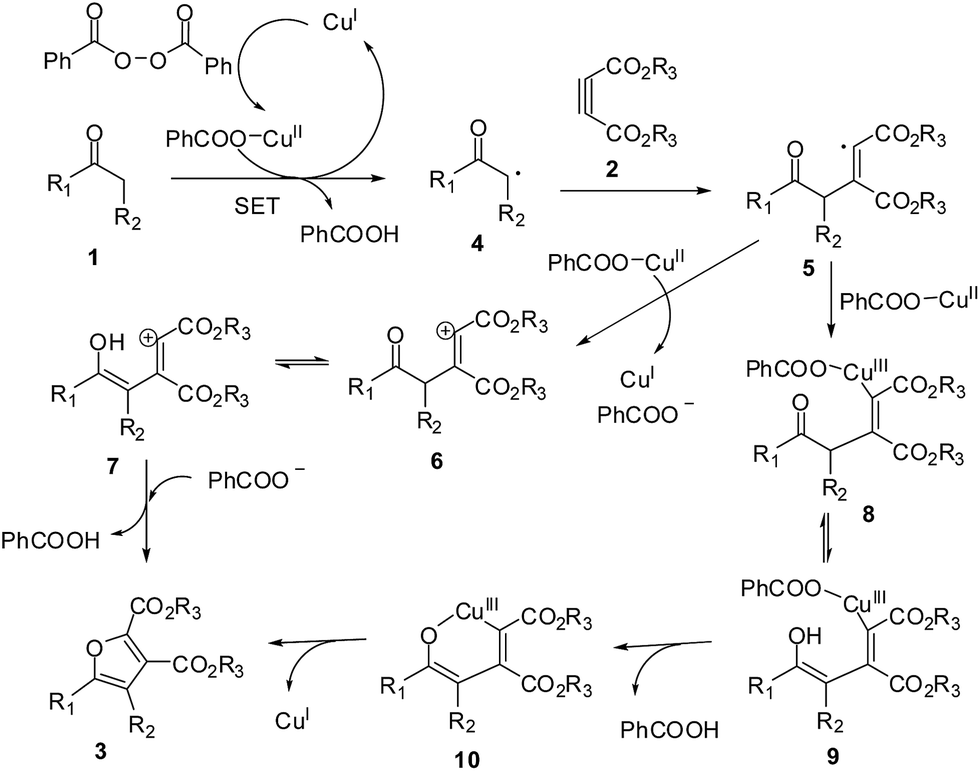
To gain further insight into the reaction mechanism, we added the radical scavenger 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) to the model reaction system. While the product (3aa) was not obtained, which indicated that a free radical pathway might be involved. The mechanism of the copper catalyzed furan synthesis has been proposed by previous related literature.10,13 One common process is the CuII/CuI catalytic cycle initiated via one-electron oxidation by CuII (single electron transfer, SET) (Scheme 4). The combination of one- and two-electron processes was also proposed to occur via a CuI/CuII/CuIII catalytic cycle, in which the key step is the formation of an organocopperIII intermediate (Scheme 4). In addition, the plausible mechanism of cyclization of 1,3-dicarbonyl compound and β-ketoester with alkynoate is similar to the related literature.15
 | ||
| Scheme 4 Plausible reaction mechanism. | ||
Conclusions
In conclusion, we have disclosed the annulation method of ketones with alkynoate catalyzed by CuI. The developed reaction is highly practical, because diverse, easily available starting materials can be used without preliminary functionalization. Also, this undecorated cyclization protocol is characterized by mild conditions, inexpensive catalyst, wide substrate scope and large-scale processes, as well as no any ligand and additive, allowing an environmentally and efficient access to biologically important scaffolds.Experimental
Unless otherwise noted, all materials were obtained from commercial suppliers and dried and purified by standard procedures. The melting point was measured on a SGW X-4 monocular microscope melting point apparatus with thermometer unadjusted. 1H NMR and 13C NMR spectra were acquired on a Bruker Avance III 400 MHz spectrometer with CDCl3 as the solvent and tetramethylsilane (TMS) as the internal standard. The chemical shifts were reported in δ (ppm). Mass spectra (MS) data were obtained using Esquire 6000 Mass Spectrometer. HRMS were obtained by ESI on a TOF mass analyzer. Column chromatography was performed with silica gel (200–300 mesh, Qingdao Haiyang Chemical Co., Ltd, China). Petroleum ether used for column-chromatography has a boiling range of 60–90 °C.Caution! Organic peroxides can be severe fire and explosion hazards.
General methods for preparing compounds 3
A 25 mL Schlenk tube was charged with various ketone (1) (0.5 mmol), alkynoate (2) (1 mmol), CuI (0.1 mmol), BPO (1 mmol) and CH3CN (2 mL). The tube was sealed, and then the mixture was stirred under air at 100 °C for 10 h. After cooling, the solvent was diluted with water (5 mL) and extracted with dichloromethane (3 × 4 mL). The combined organic layers were dried over anhydrous Na2SO4 and concentrated by a rotary evaporator, and the residue was purified by column chromatography on silica gel (petroleum ether/ethyl acetate 4:1) to provide the desired products (3).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 3.90 (s, 3H, OCH3), 3.76 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3) δ: 163.8, 163.2, 161.7, 147.0, 134.1, 130.5, 128.7, 128.2, 117.2, 53.3, 52.1; MS (ESI) m/z: 286.8 [M + Na]+, 550.9 [2M + Na]+; HRMS m/z: calcd for C14H12NaO5 [M + Na]+, 283.0577; found, 283.0579.C), 3.92 (s, 3H, OCH3), 3.91 (s, 3H, ArOCH3), 3.77 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3) δ: 165.0, 134.0, 130.6, 130.5, 128.7, 117.2, 113.7, 55.5, 53.3, 52.1, 26.3; MS (ESI) m/z: 312.9 [M + Na]+, 603.0 [2M + Na]+; HRMS m/z: calcd for C15H14NaO6 [M + Na]+, 313.0683; found, 313.0687.C), 3.91 (s, 3H, OCH3), 3.76 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3) δ: 163.8, 163.2, 161.8, 147.0, 134.0, 130.5, 128.7, 128.2, 117.2, 53.3, 52.1; MS (ESI) m/z: 304.1 [M − H]−; HRMS m/z: calcd for C14H11NNaO7 [M + Na]+, 328.0428; found, 328.0430.C), 3.91 (s, 3H, OCH3), 3.76 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3) δ: 164.0, 163.2, 162.0, 147.0, 134.0, 130.5, 128.7, 128.2, 117.2, 53.3, 52.1; MS (ESI) m/z: 304.1 [M − H]−; HRMS m/z: calcd for C14H11NNaO7 [M + Na]+, 328.0428; found, 328.0431.C), 3.90 (s, 3H, OCH3), 3.76 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3) δ:164.0, 163.2, 161.7, 147.0, 134.0, 130.5, 128.7, 128.3, 117.2, 53.3, 52.1; MS (ESI) m/z: 304.1 [M − H]−; HRMS m/z: calcd for C14H11NNaO7 [M + Na]+, 328.0428; found, 328.0429.C), 3.77 (s, 3H, OCH3), 3.63 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3) δ: 163.8, 163.3, 161.8, 147.0, 134.1, 133.9, 130.5, 130.2, 129.4, 128.7, 128.5, 124.8, 117.5, 53.3, 52.2; HRMS m/z: calcd for C15H11F3NaO5 [M + Na]+, 351.0456; found, 351.0450.C), 3.90 (s, 3H, OCH3), 3.76 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3) δ: 163.8, 163.2, 161.8, 147.0, 134.1, 130.5, 128.7, 128.2, 117.2, 53.3, 52.1; MS (ESI) m/z: 316.9 [M + Na]+, 611.0 [2M + Na]+; HRMS m/z: calcd for C14H11ClNaO5 [M + Na]+, 317.0187; found, 317.0190.C), 3.86 (s, 3H, OCH3), 3.72 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3) δ: 162.9, 161.5, 161.2, 146.9, 134.0, 130.5, 128.6, 128.3, 117.5, 62.6, 61.2; HRMS m/z: calcd for C14H11ClNaO5 [M + Na]+, 317.0193; found, 317.0190.C), 3.92 (s, 3H, OCH3), 3.77 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3) δ: 164.0, 163.2, 161.8, 147.0, 134.1, 130.5, 128.7, 128.2, 117.2, 52.3, 52.1; MS (ESI) m/z: 300.9 [M + Na]+, 579.1 [2M + Na]+; HRMS m/z: calcd for C14H11FNaO5 [M + Na]+, 301.0483; found, 301.0485.C), 3.91 (s, 3H, OCH3), 3.77 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3) δ: 163.8, 163.3, 161.8, 147.0, 134.1, 133.8, 130.5, 130.2, 128.7, 128.5, 117.2, 53.3, 52.2; MS (ESI) m/z: 332.8 [M + Na]+, 642.9 [2M + Na]+; HRMS m/z: calcd for C18H14NaO5 [M + Na]+, 333.0733; found, 333.0735.C), 3.95 (s, 3H, OCH3), 3.88 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3) δ: 171.9, 163.8, 163.3, 161.8, 147.0, 134.1, 133.8, 130.5, 130.2, 128.7, 128.5, 117.2, 53.3, 52.2; MS (ESI) m/z: 360.9 [M + Na]+, 699.0 [2M + Na]+.C), 4.32 (q, 2H, J = 6.8 Hz, OCH2CH3), 4.18 (q, 2H, J = 6.8 Hz, OCH2CH3), 1.32 (t, 3H, J = 6.8 Hz, OCH2CH3), 1.18 (t, 3H, J = 6.8 Hz, OCH2CH3); 13C NMR (100 MHz, CDCl3) δ: 163.8, 163.0, 161.2, 146.9, 134.0, 130.5, 128.6, 128.3, 117.5, 62.6, 61.2, 14.0; HRMS m/z: calcd for C16H15ClNaO5 [M + Na]+, 345.0506; found, 345.0501.C), 4.32 (q, 2H, J = 6.8 Hz, OCH2CH3), 4.18 (q, 2H, J = 7.2 Hz, OCH2CH3), 1.32 (t, 3H, J = 6.8 Hz, OCH2CH3), 1.18 (t, 3H, J = 7.2 Hz, OCH2CH3); 13C NMR (100 MHz, CDCl3) δ: 163.8, 163.3, 161.8, 147.0, 134.1, 133.8, 130.5, 130.2, 128.7, 128.5, 124.4, 117.2, 53.3, 52.2, 14.6, 14.4; HRMS m/z: calcd for C17H15F3NaO5 [M + Na]+, 379.0769; found, 379.0765.
C), 3.90 (s, 3H, OCH3), 3.76 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3) δ: 163.8, 163.2, 161.7, 147.0, 134.1, 130.5, 128.7, 128.2, 117.2, 53.3, 52.1; MS (ESI) m/z: 286.8 [M + Na]+, 550.9 [2M + Na]+; HRMS m/z: calcd for C14H12NaO5 [M + Na]+, 283.0577; found, 283.0579.C), 3.92 (s, 3H, OCH3), 3.91 (s, 3H, ArOCH3), 3.77 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3) δ: 165.0, 134.0, 130.6, 130.5, 128.7, 117.2, 113.7, 55.5, 53.3, 52.1, 26.3; MS (ESI) m/z: 312.9 [M + Na]+, 603.0 [2M + Na]+; HRMS m/z: calcd for C15H14NaO6 [M + Na]+, 313.0683; found, 313.0687.C), 3.91 (s, 3H, OCH3), 3.76 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3) δ: 163.8, 163.2, 161.8, 147.0, 134.0, 130.5, 128.7, 128.2, 117.2, 53.3, 52.1; MS (ESI) m/z: 304.1 [M − H]−; HRMS m/z: calcd for C14H11NNaO7 [M + Na]+, 328.0428; found, 328.0430.C), 3.91 (s, 3H, OCH3), 3.76 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3) δ: 164.0, 163.2, 162.0, 147.0, 134.0, 130.5, 128.7, 128.2, 117.2, 53.3, 52.1; MS (ESI) m/z: 304.1 [M − H]−; HRMS m/z: calcd for C14H11NNaO7 [M + Na]+, 328.0428; found, 328.0431.C), 3.90 (s, 3H, OCH3), 3.76 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3) δ:164.0, 163.2, 161.7, 147.0, 134.0, 130.5, 128.7, 128.3, 117.2, 53.3, 52.1; MS (ESI) m/z: 304.1 [M − H]−; HRMS m/z: calcd for C14H11NNaO7 [M + Na]+, 328.0428; found, 328.0429.C), 3.77 (s, 3H, OCH3), 3.63 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3) δ: 163.8, 163.3, 161.8, 147.0, 134.1, 133.9, 130.5, 130.2, 129.4, 128.7, 128.5, 124.8, 117.5, 53.3, 52.2; HRMS m/z: calcd for C15H11F3NaO5 [M + Na]+, 351.0456; found, 351.0450.C), 3.90 (s, 3H, OCH3), 3.76 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3) δ: 163.8, 163.2, 161.8, 147.0, 134.1, 130.5, 128.7, 128.2, 117.2, 53.3, 52.1; MS (ESI) m/z: 316.9 [M + Na]+, 611.0 [2M + Na]+; HRMS m/z: calcd for C14H11ClNaO5 [M + Na]+, 317.0187; found, 317.0190.C), 3.86 (s, 3H, OCH3), 3.72 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3) δ: 162.9, 161.5, 161.2, 146.9, 134.0, 130.5, 128.6, 128.3, 117.5, 62.6, 61.2; HRMS m/z: calcd for C14H11ClNaO5 [M + Na]+, 317.0193; found, 317.0190.C), 3.92 (s, 3H, OCH3), 3.77 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3) δ: 164.0, 163.2, 161.8, 147.0, 134.1, 130.5, 128.7, 128.2, 117.2, 52.3, 52.1; MS (ESI) m/z: 300.9 [M + Na]+, 579.1 [2M + Na]+; HRMS m/z: calcd for C14H11FNaO5 [M + Na]+, 301.0483; found, 301.0485.C), 3.91 (s, 3H, OCH3), 3.77 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3) δ: 163.8, 163.3, 161.8, 147.0, 134.1, 133.8, 130.5, 130.2, 128.7, 128.5, 117.2, 53.3, 52.2; MS (ESI) m/z: 332.8 [M + Na]+, 642.9 [2M + Na]+; HRMS m/z: calcd for C18H14NaO5 [M + Na]+, 333.0733; found, 333.0735.C), 3.95 (s, 3H, OCH3), 3.88 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3) δ: 171.9, 163.8, 163.3, 161.8, 147.0, 134.1, 133.8, 130.5, 130.2, 128.7, 128.5, 117.2, 53.3, 52.2; MS (ESI) m/z: 360.9 [M + Na]+, 699.0 [2M + Na]+.C), 4.32 (q, 2H, J = 6.8 Hz, OCH2CH3), 4.18 (q, 2H, J = 6.8 Hz, OCH2CH3), 1.32 (t, 3H, J = 6.8 Hz, OCH2CH3), 1.18 (t, 3H, J = 6.8 Hz, OCH2CH3); 13C NMR (100 MHz, CDCl3) δ: 163.8, 163.0, 161.2, 146.9, 134.0, 130.5, 128.6, 128.3, 117.5, 62.6, 61.2, 14.0; HRMS m/z: calcd for C16H15ClNaO5 [M + Na]+, 345.0506; found, 345.0501.C), 4.32 (q, 2H, J = 6.8 Hz, OCH2CH3), 4.18 (q, 2H, J = 7.2 Hz, OCH2CH3), 1.32 (t, 3H, J = 6.8 Hz, OCH2CH3), 1.18 (t, 3H, J = 7.2 Hz, OCH2CH3); 13C NMR (100 MHz, CDCl3) δ: 163.8, 163.3, 161.8, 147.0, 134.1, 133.8, 130.5, 130.2, 128.7, 128.5, 124.4, 117.2, 53.3, 52.2, 14.6, 14.4; HRMS m/z: calcd for C17H15F3NaO5 [M + Na]+, 379.0769; found, 379.0765.Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (21102003) and Scientific Research Foundation for the Introduction of Talent of Anhui University of Science & Technology.Notes and references
- (a) X. L. Hou, H. Y. Cheung, T. Y. Hon, P. L. Kwan, T. H. Lo, S. Y. Tong and H. N. C. Wong, Tetrahedron, 1998, 54, 1955 CrossRef CAS; (b) B. H. Lipshutz, Chem. Rev., 1986, 86, 795 CrossRef CAS; (c) M. Ghosh, S. Mishra, K. Monir and A. Hajra, Org. Biomol. Chem., 2015, 13, 309 RSC.
- (a) L. Knorr, Ber. Dtsch. Chem. Ges., 1884, 17, 2863 CrossRef; (b) C. Paal, Ber. Dtsch. Chem. Ges., 1884, 17, 2756 CrossRef.
- (a) E. Benary, Ber. Dtsch. Chem. Ges., 1911, 44, 489 CrossRef CAS; (b) F. Feist, Ber. Dtsch. Chem. Ges., 1902, 35, 1537 CrossRef CAS.
- Z. H. Gu, X. K. Wang, W. Shu and S. G. Ma, J. Am. Chem. Soc., 2007, 129, 10948 CrossRef CAS PubMed.
- B. L. Lu, J. L. Wu and N. Yoshikai, J. Am. Chem. Soc., 2014, 136, 11598 CrossRef CAS PubMed.
- Y. Z. i. Xia, A. S. Dudnik, V. Gevorgyan and Y. H. Li, J. Am. Chem. Soc., 2008, 130, 6940 CrossRef CAS PubMed.
- X. Cui, X. Xu, L. Wojtas, M. M. Kim and X. P. Zhang, J. Am. Chem. Soc., 2012, 134, 19981 CrossRef CAS PubMed.
- A. W. Sromek, A. V. Kel'in and V. Gevorgyan, Angew. Chem., Int. Ed., 2004, 43, 2280 CrossRef CAS PubMed.
- M. Zhang, H. F. Jiang, H. Neumann, M. Beller and P. H. Dixneuf, Angew. Chem., Int. Ed., 2009, 48, 1681 CrossRef CAS PubMed.
- (a) S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234 CrossRef CAS PubMed; (b) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062 CrossRef CAS PubMed; (c) X. X. Guo, D. W. Gu, Z. X. Wu and W. B. Zhang, Chem. Rev., 2015, 115, 1622 CrossRef CAS PubMed.
- W. B. Liu, H. F. Jiang, M. Zhang and C. R. Qi, J. Org. Chem., 2010, 75, 966 CrossRef CAS PubMed.
- H. Cao, H. Y. Zhan, J. H. Cen, J. G. Lin, Y. G. Lin, Q. X. Zhu, M. L. Fu and H. F. Jiang, Org. Lett., 2013, 15, 1080 CrossRef CAS PubMed.
- S. Manna and A. P. Antonchick, Org. Lett., 2015, 17, 4300 CrossRef CAS PubMed.
- During the preparation of this manuscript, Antonchick and Manna reported the direct C(sp3)–H bond functionalization annulation of acetophenone derivatives and alkyl acetylenedicarboxylates via Cu(I)-catalyzed using 2,2′-bipyridine as the ligand under radical reaction conditions. See: S. Manna and A. P. Antonchick, Org. Lett., 2015, 17, 4300 CrossRef CAS PubMed.
- R. Yan, J. Huang, J. Luo, P. Wen, G. Huang and Y. Liang, Synlett, 2010, 1071 CAS.
- (a) H. Cao, H. Y. Zhan, J. Y. Wu, H. P. Zhong, Y. G. Lin and H. Zhang, Eur. J. Org. Chem., 2012, 12, 2318 CrossRef; (b) H. Cao, H. F. Jiang, X. S. Zhou, C. R. Qi, Y. G. Lin, J. Y. Wu and Q. M. Liang, Green Chem., 2012, 14, 2710 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: NMR Spectra for products 3. See DOI: 10.1039/c5ra23058f |
| This journal is © The Royal Society of Chemistry 2016 |