
Selected organophosphorus compounds with biological activity. Applications in medicine
Sebastian Demkowicz*,
Janusz Rachon,
Mateusz Daśko and
Witold Kozak
Department of Organic Chemistry, Chemical Faculty, Gdansk University of Technology, Narutowicza 11/12, 80-233 Gdansk, Poland. E-mail: sebdemko@pg.gda.pl
First published on 12th January 2016
Abstract
The purpose of this article is to provide an overview of the latest applications of organophosphorus compounds (OPs) that exhibit biological activity. A large family of OPs have become popular in recent years. The practical application of OPs in modern medicine has been attributed to their unique properties. In this article, the methods used to select these compounds will be emphasized. This paper will first outline the findings of a literature review on OPs, including anticancer and antiviral agents, bisphosphonates, phosphorus analogues of amino acids and peptides, and organophosphorus metal complexes, and secondly, it will classify the compounds according to their biological activity and applications in the treatment of diseases.
1. Introduction
Organophosphorus compounds (OPs), which are a wide class of chemical compounds containing organic moieties usually bonded directly to phosphorus or bonded through a heteroatom, such as sulfur, oxygen or nitrogen, are some of the most common chemicals in the human environment. Because of their unique properties and high biological activity, they have largely been used worldwide in agricultural (pesticides),1 industrial (production of lubricants, hydraulic fluids, and plastics materials),2 medicinal (drugs against osteoporosis, anticancer and antiviral compounds)3,4 or veterinary (anthelmintics) applications.5 The first potent synthetic organophosphorus poison, tetraethyl pyrophosphate (TEPP), was synthesized by Clermont in 1854. At the beginning of the twentieth century, some very toxic compounds were used in many armed conflicts as chemical weapons, known as chemical warfare agents (CWA). Following the German laboratories discovery of soman, sarin and tabun, the United States and England developed VX production technologies. The book “Chemical Warfare Agents”6 discusses the physicochemical properties of chemical warfare agents, their dispersion and fate in the environment, their toxicology and management of their effects on humans, decontamination, and protective equipment. After the Second World War, OPs have been used mainly as pesticides for plants and animals. Furthermore, OPs have practically contributed to the substantial benefits for efficient food production and the fight against many serious diseases, such as malaria, yellow fever, typhus,7 or smallpox.42. Bisphosphonates in the treatment of osteoporosis
Osteoporosis is one of the most serious health problems in the world, and prevention and treatment are of great interest in the European Union, which issued “Report on Osteoporosis in the European Community – Action for Prevention”.8 The scale of the problem is alarming. One in three women and one in eight men over the age of 50 years will experience at least one fracture due to osteoporosis in their lifetime. The main criterion for selecting an osteoporosis therapy is its impact on the risk of osteoporotic fracture (femoral neck, spine, wrist). The following pharmacological methods are used:• hormone replacement therapy;
• specific estrogen receptor modulators (SERMs);
• calcitonin;
• vitamin D3 with active metabolites and calcium;
• fluorine.
Bisphosphonates are currently the most important and effective class of drugs developed for the treatment of metabolic bone disorders associated with increased osteoclast-mediated bone resorption, such as osteoporosis9,10 and Paget's disease.11,12 They are effective inhibitors of tumor-induced bone destruction and significantly reduce the incidence of skeletal complications in patients with bone metastases from several forms of cancer, including breast and prostate cancer.13 Bisphosphonates have a high affinity for calcium and therefore specifically target bone mineral, where they are internalized by bone-destroying osteoclasts and inhibit their function.14 Importantly, potential of bisphosphonates has also been identified in areas ranging from parasite-growth inhibition to immunological and cancer therapeutics.3 These compounds primarily affect the function of osteoclasts, but recent preclinical evidence indicates that other neighboring cell types, such as macrophages, monocytes and cancer cells, could also be targets for these drugs.15 Bisphosphonates have been shown to induce tumor cell apoptosis, to modulate cells in the immune system and to inhibit tumor angiogenesis.
Bisphosphonates used in clinical practice are characterized by a P–C–P structure.16 This structure allows for a wide variety of variations. The first bisphosphonates were synthesized by German chemists and were first used only for a few industrial applications, such as antiscaling agents. It was only in 1968–1969 that H. Fleisch et al. demonstrated that these compounds had biological effects, specifically on calcified tissues.17–20 Replacing the O atom with a CH2 group in the pyrophosphoric acid structure created new bioactive phosphorus agents and resulted in a major breakthrough in the treatment of bone disease. It is hypothesized that bisphosphonic acid 1 are isosteric with pyrophosphoric acid 2 but are hydrolytically stable, and if attracted to bone, may block resorption since inorganic pyrophosphate inhibits the formation and dissolution of hydroxyapatite in bone (Fig. 1).
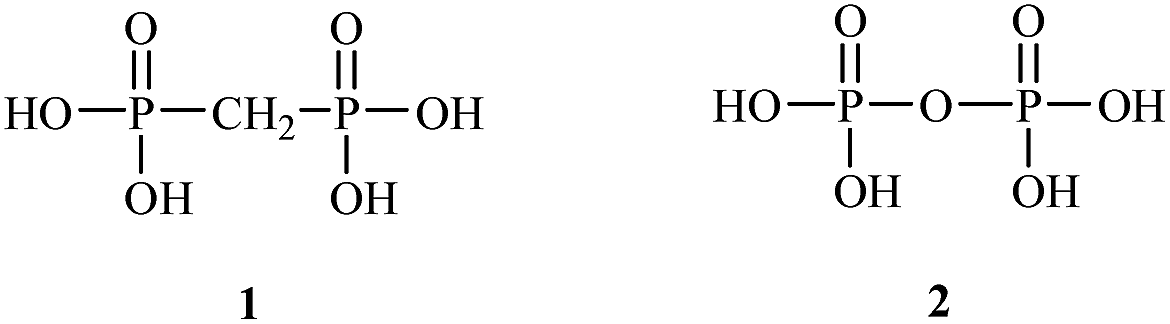 | ||
| Fig. 1 Chemical structures of bisphosphonic acid 1 and pyrophosphoric acid 2. | ||
E. Breuer's research group from the Hebrew University of Jerusalem between 1992 and 2005 had a significant impact on the synthesis and biological evaluation of bisphosphonates. E. Breuer's research group authored numerous publications and patents concerning the use of bisphosphonates for the treatment of osteoporosis. These researchers were focused on synthesizing novel bifunctional compounds, such as bisacylphosphonates 3,21 tetrakisphosphonates 4,22 bisphosphonic acid betaine derivatives 5 (ref. 23) and hydroxyiminophosphonates 6,24 and examining their effects compared to other clinically used drugs (Fig. 2).
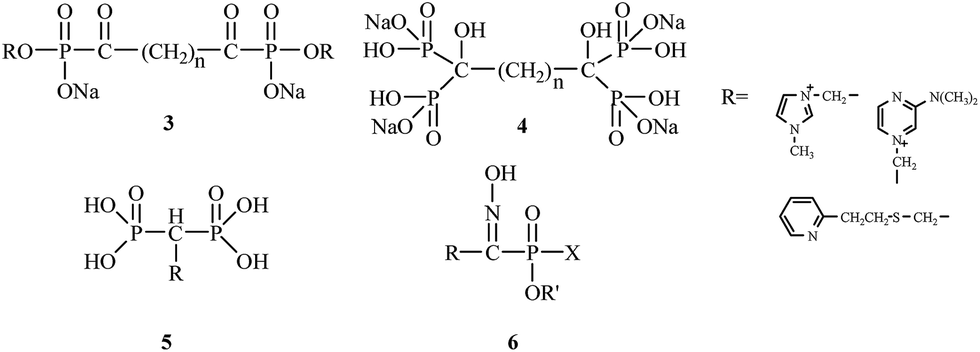 | ||
| Fig. 2 Chemical structures of bisacylphosphonates 3, tetrakisphosphonates 4, bisphosphonic acid betaine derivatives 5 and hydroxyiminophosphonates 6. | ||
The drugs clodronate 7, tiludronate 8 and etidronate 9 were among the first to be used in the clinic.
Attention has recently been drawn to the derivatives with aminoalkyl side chains for the next generation of bisphosphonates, as found in pamidronic acid 10, alendronic acid 11, olpadronic acid 12, neridronic acid 13, ibandronic acid 14 and risedronic acid 15.
The research field is currently very active, and many new discoveries in bisphosphonate bone-disease therapy have been published.25 Bisphosphonates are currently in the third generation, which includes incadronic acid 16, minodronic acid 17 and zoledronic acid 18. In addition, several bisphosphonates act as antidepressant 19 and antihypercholesterolemic agents 20 (Fig. 3).26,27
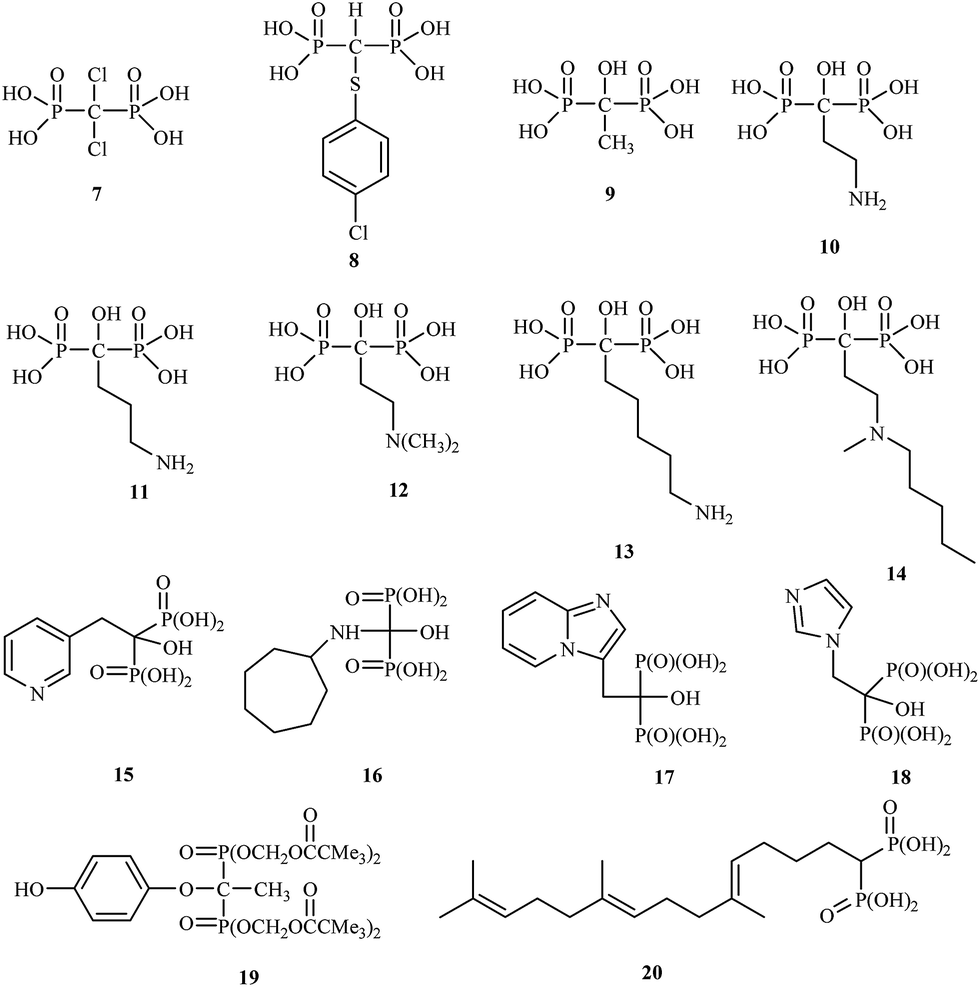 | ||
| Fig. 3 Chemical structures of bisphosphonates 7–20. | ||
3. Organophosphorus compounds as anticancer drugs
Recent research findings suggest that a number of OPs are used as anticancer drugs or have potential anticancer properties. They are usually used in oncology as alkylating chemotherapeutic agents. These compounds react with DNA, RNA and some enzymes. N,N′,N′′-Triethylenethiophosphoramide 21, sold under the trade name thioTEPA, is a compound used as an anticancer chemotherapeutic drug that binds to DNA, crosslinks the two strands and prevents cell duplication. N,N′,N′′-Triethylenethiophosphoramide, developed in the 1950s, is a trifunctional alkylating agent with a broad spectrum of antitumor activity.28 This drug is used to treat many diseases, including breast cancer, ovarian cancer, lymphosarcoma, superficial papillary carcinoma of the urinary bladder, and Hodgkin's disease (Fig. 4).29 | ||
| Fig. 4 Chemical structures of organophosphorus alkylating agents 21–26. | ||
Several derivatives of N,N′,N′′-triethylenethiophosphoramide have been synthesized, and their antitumor activity was evaluated. McCracken and co-workers carried out the synthesis of N-[bis(1-aziridinyl)phosphoro]-carbamate 22.30 In vivo studies demonstrated that the synthesized drugs had a higher toxicity against cancer cells, and they did not invade other tissues. In 1963, Chernov et al.31 published the synthesis and biological evaluation of phosphazine 23. Chernov's research demonstrated phosphazine's high activity 23 against transplanted carcinoma in mice, rats, and rabbits. According to these investigations, phosphazine is toxic; however, its toxicity is much lower than thio-phosphamide (thio-TEPA) or dipin (N,N′-bis(diaziridinylphosphinylidyne)piperazine). In 1968, Noell et al.32 synthesized methylphosphazine (P,P-bis(2-methyl-1-aziridinyl)-N-2-pyrimidinylphosphosphinic amide) 24, which demonstrated excellent activity against both leukemia L1210 and Walker 256 and was more active and less toxic than several clinical drugs, such as thioTEPA and phosphazine. In the 1980s, further structural modifications were tested. Sosnovsky synthesized the nitroxyl-labeled analogues of tiamide (TEPA).33 These studies showed that compounds 25 and 26 possess therapeutic indexes 8- to 12-fold higher than those of thio-TEPA and TEPA.
Several of the most important OPs that exhibit anticancer properties are oxazaphosphorines. Even today, 50 years after its introduction, this class of compounds is one of the most widely used cytostatics. The more than 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 scientific publications regarding this compound demonstrate the wide interest it has received.
000 scientific publications regarding this compound demonstrate the wide interest it has received.
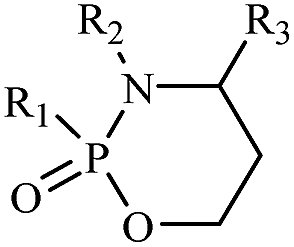
These compounds (Table 1), classified as alkylating agents, are therapeutically inactive prodrugs that must be activated to induce their cytotoxicity.35,36 Currently, cyclophosphamide 27 and ifosfamide 29 are used commonly for the treatment of non-Hodgkin lymphomas and a variety of bone and soft tissue sarcomas.37 Compared to many other anticancer drugs, cyclophosphamide exhibits relatively little non-hematopoietic toxicity.
|
|||
|---|---|---|---|
| Substance | R1 | R2 | R3 |
| Cyclophosphamide 27 | 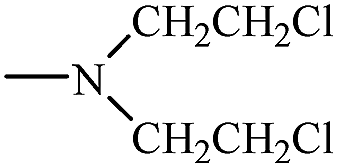 |
H– | H– |
| Mafosfamide 28 | 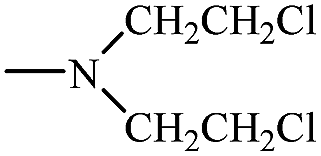 |
H– | −O3S–H2C–CH2–S– |
| Ifosfamide 29 | 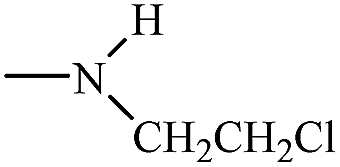 |
Cl–H2C–CH2– | H– |
| Trofosfamide 30 |  |
Cl–H2C–CH2– | H– |
In 2000, a series of naphthoquinone 31 and benzimidazolequinone 32 phosphorodiamidates were synthesized and studied as potential cytotoxic prodrugs activated by DT-diaphorase,38 and an activation process was proposed (Fig. 5). To activate the synthesized prodrugs, the quinone moiety is reduced to form phosphoramide mustard. All compounds were excellent substrates for human DT-diaphorase. The naphthoquinones 31 showed high toxicity towards both HT-29 and BE human colon cancer cell lines and were 1- to 2-fold more active than the benzimidazolequinone derivatives 32.
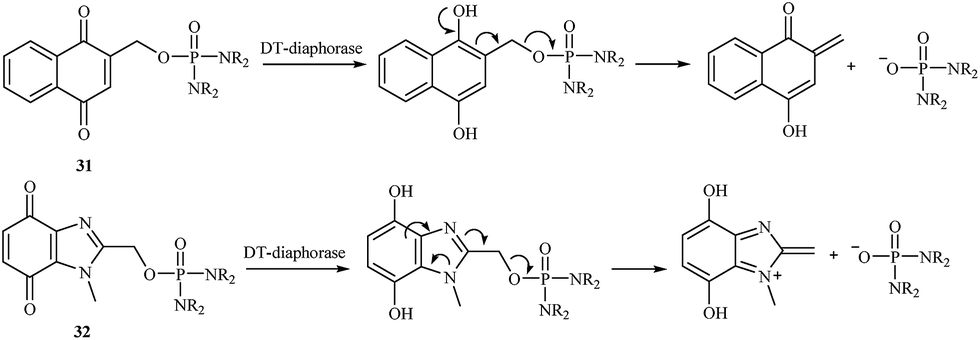 | ||
| Fig. 5 Activation process of naphthoquinone 31 and benzimidazolequinone 32 phosphorodiamidates. | ||
In 2008, Jian-Xin Duan et al. synthesized new phosphoroorganic compounds with anticancer activity based on 2-nitroimidazole derivatives.39 These compounds are hypoxia-activated achiral phosphoramidate mustards synthesized based on the DNA cross-linking toxin from the prodrug ifosfamide. Hypoxia-activated phosphoramidates were introduced by Borch and coworkers.40 The most successful were the 5-nitrothiophene- and 5-nitrofuran-triggered prodrugs of phosphoramidate toxins. The synthetic methods (Fig. 6) were very straightforward and high yielding. The most promising antitumor agent was TH-302 33, which demonstrated excellent in vivo efficacy and is currently in clinical trials.
 | ||
| Fig. 6 Synthesis of TH-302 33. | ||
In 2003, Jain et al. proposed a new class of 1,2-benzisoxazole phosphorodiamidate compounds.41 As expected, in vivo studies showed that the compounds 34, 35, and 36 were 3- to 5-fold more active than analogues 37 and 38 (Fig. 7).
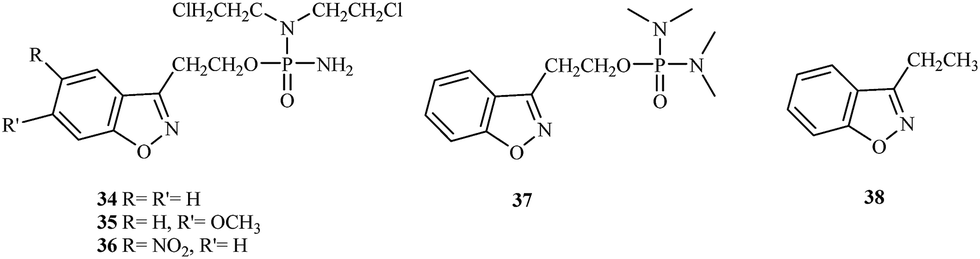 | ||
| Fig. 7 Structures of benzisoxazole phosphorodiamidates 34–37 and reference compound 38. | ||
Organophosphorus compounds are also used as prospective treatments for hormone-dependent breast cancer. They are included in the list of steroid sulfatase (STS) inhibitors, an enzyme involved in the biosynthesis of estrogen in the mammary glands. The WHO lists estrogen as an important factor in the development of breast cancer. Breast cancer is the most frequently diagnosed cancer in the female population of industrialized countries. Estimates for 2014 show more than 230000 diagnosed cases of breast cancer and more than 40000 deaths (according to National Cancer Institute data). Inhibitors based on OPs show potential as new breast cancer therapies because they are structurally similar to the natural steroid sulfatase substrate and have excellent binding affinities to the enzyme active site.42
The initial strategy employed for generating a lead STS inhibitor involved replacement of the sulfate group (OSO3) on the natural enzyme substrate with surrogates or mimics such as phosphates or thiophosphates. Recently, Demkowicz et al. synthesized new phosphate and thiophosphate esters of tricyclic coumarin 39,43,44 N-alkanoyl tyramine 40,45 biphenyl 41 (ref. 46) and flavone 42 (ref. 47) derivatives as potent STS inhibitors. The most active compound, 4-(2-dodecanoylamino-ethyl)-phenyl dimethyl phosphate, demonstrated the greatest inhibitory effect, with IC50 values of 390 nM in enzymatic assay with STS isolated from the human placenta. Although the mechanism of activity is unknown, docking studies conducted to explore the potential interactions between synthesized compounds and the active site of STS suggest a phosphate group transfer to one of the key amino acid residue involved in the enzymatic reaction (FGly75) or methylation of this residue during the inactivation process.
In 2015, the same research group synthesized a series of bicoumarin 43 (ref. 48) and biflavone 44 (ref. 47) thiophosphate derivatives as STS inhibitors. The most active compound, bis-(6-oxo-7,8,9,10-tetrahydro-6H-benzo[c]chromen-3-yl) hydrogenthiophosphate, inhibit STS activity with IC50 values of 860 nM in enzymatic assay with purified STS. Although the mechanism of inhibition also remains unclear, molecular modeling suggests a completely different manner of binding to the active site of STS in comparison to previous organophosphorus inhibitors of STS. Indeed, they can adopt conformations that are able to fill the whole cavity and prevent the substrate's access to the catalytic amino acid residues.
In 2001, David et al. demonstrated the utility of a new anticancer drug called apomine 45, a per os-active, apoptosis-inducing agent that recently entered clinical trials in cancer patients.49 The clinical trial findings revealed that the drug can selectively inhibit cell proliferation and induce tumor cell apoptosis through the farnesoid X receptor. In vitro assay results showed that 63 and 91% of ovarian cancers were sensitive to apomine at concentrations of 10 and 20 μM, respectively. This compound also inhibits the mevalonate/isoprenoid pathway of cholesterol synthesis and may also prove effective as a skin cancer chemoprevention therapy.50
Another drug used in cancer therapy is combretastatin A-4 phosphate (CA4P) 46.51 Combretastatin A-4 phosphate is a novel microtubule destabilizing drug, a type of vascular-targeting agent designed to damage the vasculature (blood vessels) of cancer tumors and cause central necrosis. It is the first of a series of combretastatin analogs to enter the clinic (Fig. 8).
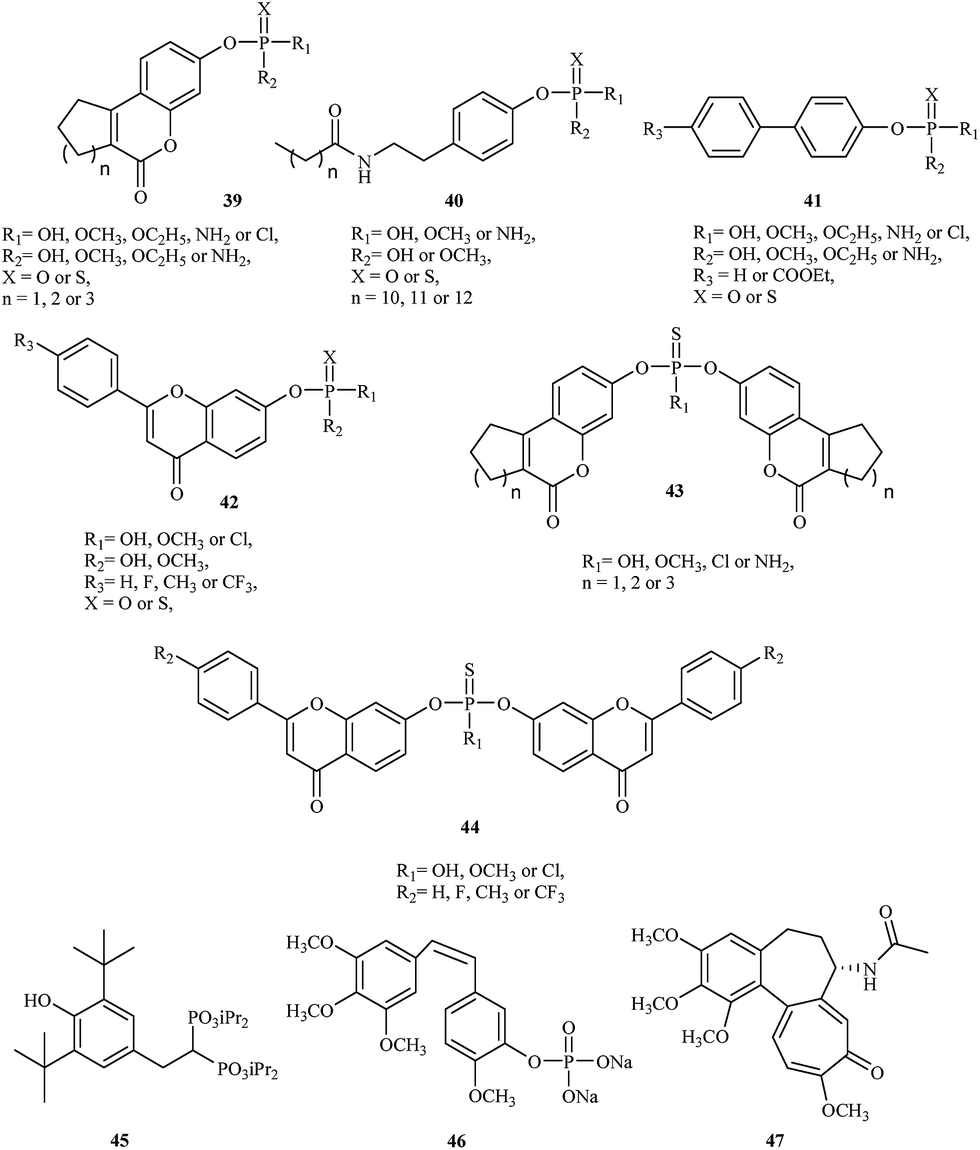 | ||
| Fig. 8 Chemical structures of organophosphorus anticancer agents 39–47. | ||
The structure of CA4P is similar to colchicine 47, and it binds the colchicine-binding site on tubulin and inhibits tubulin polymerization.52 Clinical studies showed that CA4P's toxicity profile is consistent with a drug that is vascularly active and devoid of traditional cytotoxic side effects.49
The latest achievement in the treatment of cancer is the use of antisense drugs.53–55 Antisense therapy is based on oligonucleotides expected to stop or reduce the expression of selected genes using different approaches based on sequence specific targeting of nucleic acids.56 This new anticancer strategy has been widely used to treat cancers such as colorectal carcinoma, lung cancer, pancreatic carcinoma, malignant glioma or malignant melanoma. Most therapies have not yet produced significant clinical results, and application of this method is the subject of intensive investigations.
4. Organophosphorus metal complexes
Research about the applications of metal complexes in medicine is one of the most integrated areas of science, combining data regarding structure, properties of metal complexes, and control of the body's vital processes. The design of new drugs based on metal complexes is an important contribution to the development of more efficient chemotherapy methods. These metal complexes are used in the treatment of many diseases. However, it was not until the early 1960's that the biological effect of cisplatin was discovered.57 Primary research by Rosenberg indicated that metal ions were capable of binding to nucleic acids, thereby altering their conformation and biological function.58 Metal complexes play an important role in many biological processes59 including cell division and gene expression, as well as processes such as carcinogenicity or toxicity.60 Among the synthesized compounds, many very important complexes are based on OPs. The literature indicates that most studies consider OPs to have potential anti-cancer aspects. Their activity against many types of cancer is currently the subject of intensive research. Organophosphorus metal complexes include platinum, ruthenium, palladium, gold and copper metal centers.High in vitro activities of ruthenium complexes against different cancer cell lines suggest that they are the most promising in anticancer therapy and may play a dominant role as antitumor drugs compared to others metal complexes.61 Among, a number of ruthenium complexes, NAMI-A and KP1019 are currently entered into a clinical trials.62,63 Although, the mechanism of action for these compounds is not fully understood,64 recent reports indicate that antiproliferative activity of Ru complexes is strengthened with the interactions with DNA and different cellular proteins.65 Among the many synthesized ruthenium compounds that exhibit antitumor activity, the arene PTA ruthenium(II) complexes (RAPTA) 48–56 are of great interest (Fig. 9).66 The RAPTA complexes 48–56 were found to exhibit pH-dependent DNA damage. Although the cytotoxicity of the new complexes proved to be lower compared to cisplatin, progress has been made in designing new and selective drugs without the harsh side effects of cisplatin.
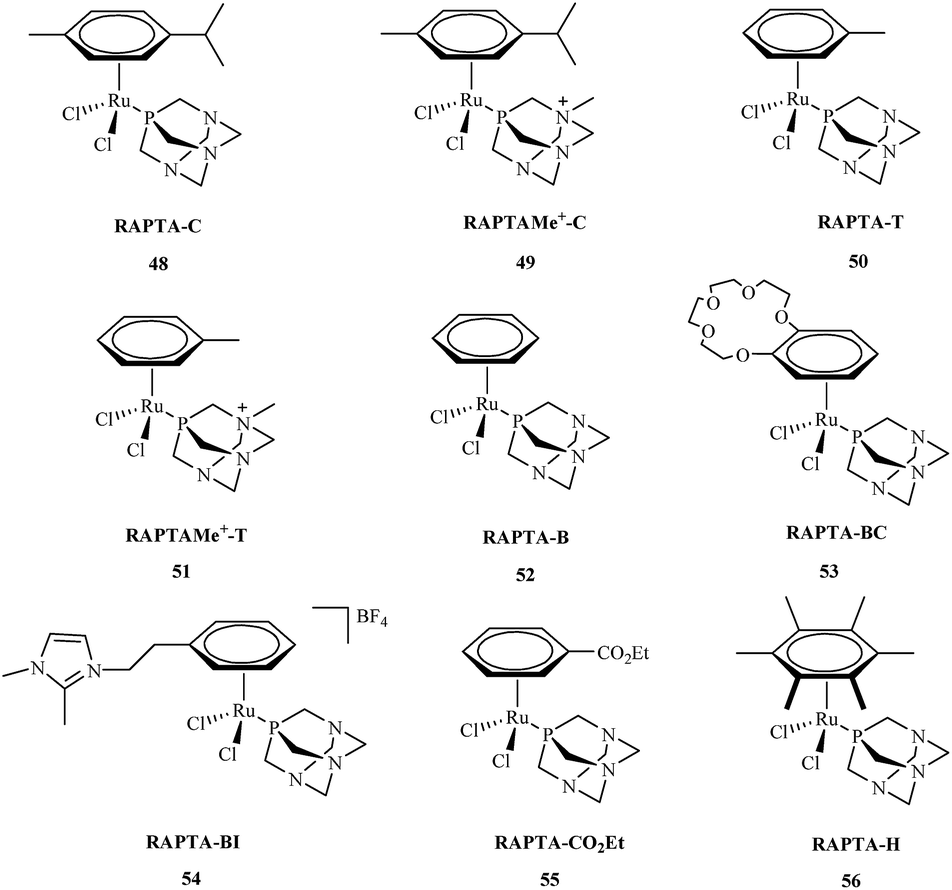 | ||
| Fig. 9 Chemical structures of RAPTA complexes 48–56. | ||
Efforts to synthesise of ruthenium complexes that exhibit potent anticancer properties led to the discovery of other RAPTA derivatives. [Ru(η6-p-phenylethacrynate)Cl2(pta)] – ethaRAPTA 57, a derivative of RAPTA-C 48 is one of the most promising (Fig. 10). Chakree et al. characterized the ethaRAPTA 57 interactions with DNA and found out that ethaRAPTA exposed a higher efficiency in comparison to RAPTA-C in inhibiting the breast cancer suppressor gene 1.67
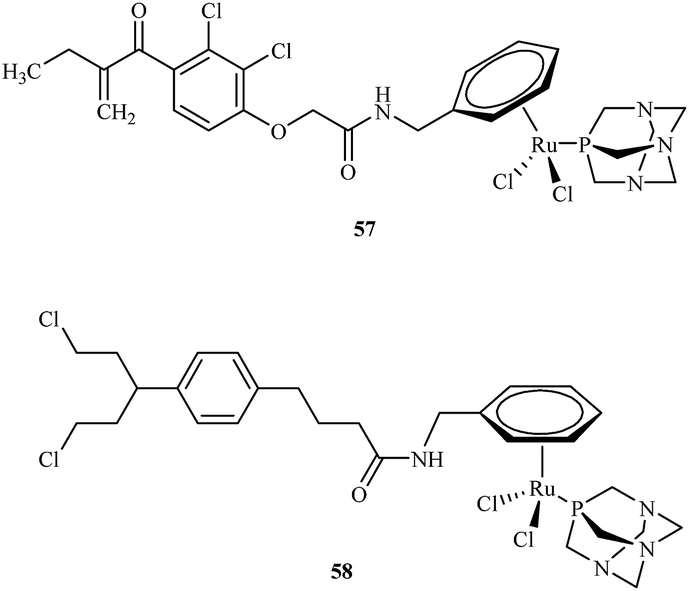 | ||
| Fig. 10 Chemical structures of ethaRAPTA 57 and 58 complexes. | ||
Recent advances in RAPTA complexes developments led to synthesis of compounds that were the modification of RAPTA scaffold with chlorambucil.68 Chlorambucil is well known DNA alkylating agent whereas RAPTA complexes coordinate to amino acid residues of proteins. Biological activities of tested RAPTA derivatives were in the low μM range against A2780, A2780R and MCF-7 cell lines. Among a series of newly obtained RAPTA complexes, the most potent was 58 with IC50 values for A2780, A2780R and MCF-7 cell lines of 8.3, 10.0 and 12.0 μM, respectively.
Many metal complexes with triphenylphosphine and other tertiary phosphines have been reported to be catalysts for various processes, such as polymerization of alkenes and acetylenes, Wilkinson catalyst,69 oxo hydroformylation of alkenes with hydrogen and CO,70 asymmetric Pauson–Khand71 and Morita–Baylis–Hillman72 reactions, synthesis of enantiomerically enriched cyclohexadiene by reaction of terminal dieneyne using a chiral iridium complex73 and asymmetric allylation and propargylation of ketones.74 Many phosphines and diphosphines are optically active due to an asymmetric phosphorus or a carbon atom and have been used to for asymmetric hydrogenations.75
5. Phosphorus analogs of amino acids and peptides
Aminophosphinic and aminophosphonic acids are a very important group of chemical compounds, and their synthesis has attracted considerable attention in medicinal chemistry. Although the biological importance of these compounds was discovered in the 1950's, they still represent a promising class of potential drugs. They are classified as antimetabolites, which compete with their aminocarboxylic acid analogues in the active sites of enzymes and other cell receptors.76–795.1. Aminophosphinic acid derivatives
V. Dive synthesized and evaluated a series of cyclic peptides containing a phosphinic bond as potential zinc bacterial collagenase inhibitors.80 Studying collagenases or other proteases helps to better understand the pathology and treatment of human diseases such as rheumatoid arthritis, tissue repair, metastasis, angiogenesis or cirrhosis. Among the synthesized pseudopeptides with different sized cyclic rings, they found two compounds, (cyclo[Gly-Pro-Phe-ψ(PO2CH2)-Gly-Pro-Ahx]) and (cyclo[βAla-Pro-Pheψ(PO2CH2) Gly-Pro-Ahx]), exhibited very potent inhibition activity with Ki values of 120 and 90 nM, respectively. The authors found that the stereochemistry and conformation of the pseudophenylalanine residue determined the potency of these cyclic peptides.Bartlett and coworkers reported the synthesis and application of phosphinic acid peptide analogues, potent slow-binding inhibitors of aspartic peptidases.81 They showed that incorporation of a phosphorus-containing analogue of the amino acid statine into the oligopeptide sequences significantly increased inhibition of the prototypical aspartic peptidase pepsin. During the course of the investigations, they discovered that the more effective inhibitor for each pair of the diastereomers was the L-configuration (Table 2).
| Inhibitor | Ki |
|---|---|
| Iva-D-StaP-Ala-Iaa 59A | 25 μM |
| Iva-L-StaP-Ala-Iaa 59B | 0.9 μM |
| Iva-Val-D-StaP-Ala-Iaa 60A | 200 nM |
| Iva-Val-L-StaP-Ala-Iaa 60B | <0.07 nM |
Several aminophosphonic acids were found to be very selective and potent inhibitors toward other endopeptidases. B. Vincent's research group published the biological studies of a series of selective and potent phosphinic peptide inhibitors of endopeptidase 3.4.24.16.82 This research showed that the most selective peptide analog, Pro-Phe-ψ(PO2CH2)-Leu-Pro-NH2, displayed a Ki value of 12 nM. In comparison to the related endopeptidase 3.4.24.15 tested, the inhibitor was 5540-fold less effective; furthermore, compared to other enzymes (endopeptidase 3.4.24.11, aminopeptidases B and M, dipeptidylaminopeptidase IV or proline endopeptidase), no inhibition was observed.
Phosphinic acid derivatives have also been utilized as inhibitors of metalloproteinases. Matrix metalloproteinases (MMPs) are zinc-dependent proteolytic enzymes. They contain a zinc cation (Zn2+) that plays both a catalytic and structural role.83 There are over 30 types of metalloproteinases involved in many physiological and pathological remodeling and degradation processes of extracellular matrix components.84 In many cases, a change in MMP function causes a pathological state; they are also implicated in many diseases including cancer,85 osteoporosis,86 arthritis,87 arteriosclerosis,88 multiple sclerosis,89 and liver cirrhosis.90 Fosinopril 61 was one of the first phosphorus-based MMP inhibitors clinically used for the treatment of hypertension and some types of chronic heart failure by inhibition of ACE (angiotensin converting enzyme).91 Fosinopril is administered as a prodrug and converted in vivo to the active form fosinoprilat 62. Direct administration of the drug in its active form does not guarantee effective treatment because fosinoprilat is ionic under physiological conditions, and its oral bioavailability is thus very low.
In 1999, Vassiliou and coworkers reported the synthesis and biological studies of a series of phosphinic pseudo-tripeptide inhibitors against MMP-1, MMP-2, MMP-7, MMP-8, MMP-11, and MMP-14.92 They demonstrated the structure–activity relationships regarding the influence of different substituents at the P1′, P2 and P2′ position. The inhibitors were highly potent towards MMPs, displaying nanomolar Ki values. For MMP-8, replacement of the benzyl group at the P1′ position by phenylpropyl (RXP03) 63 caused a 30-fold increase in potency (Fig. 11).
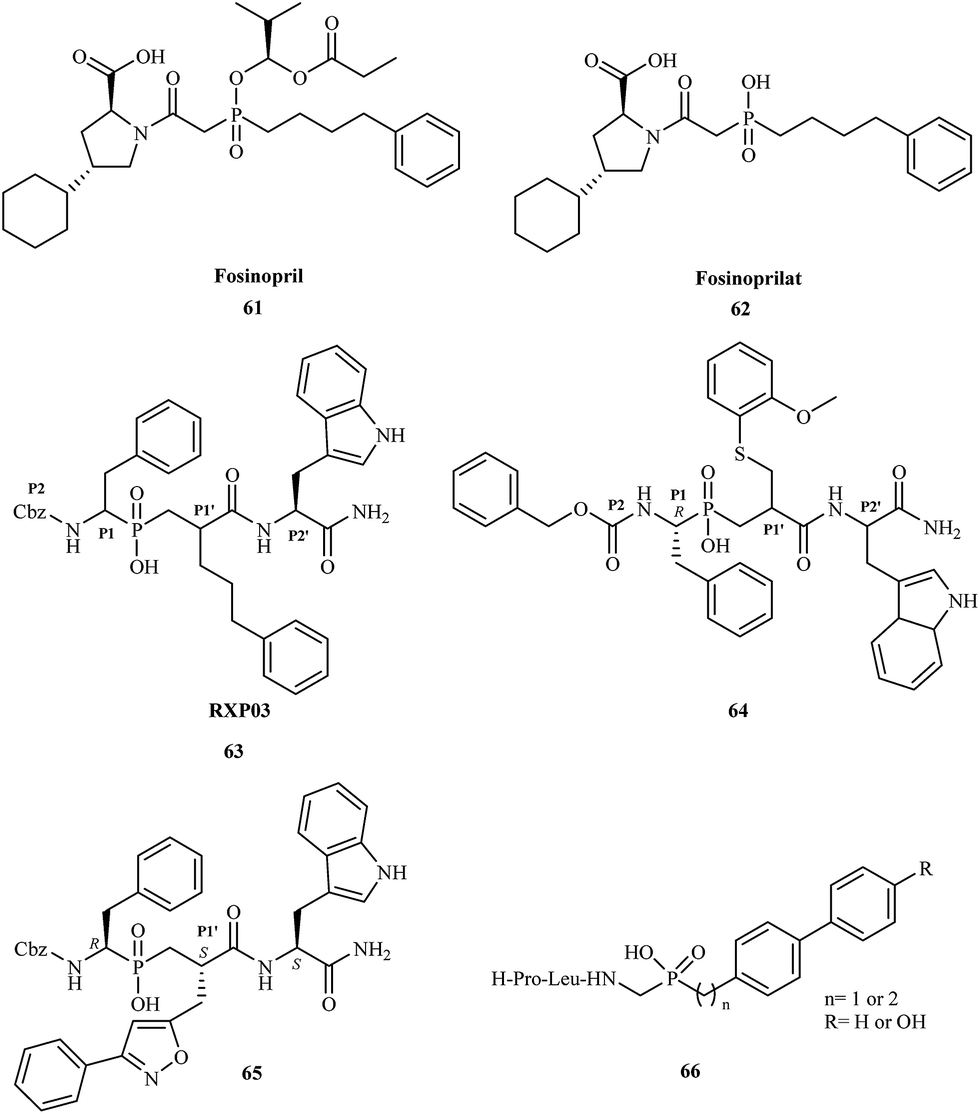 | ||
| Fig. 11 Chemical structures of aminophosphinic acid derivatives 61–66. | ||
Yiotakis and coworkers published the synthesis of a phosphinic pseudopeptide series containing a variety of P1-side chains.93 The new compounds were tested as potential selective inhibitors against MMP-2, MMP-7, MMP-8, MMP-9, MMP-11, MMP-13, and MMP-14. Several phosphinic inhibitors displayed high selectivity toward MMP-11; the greatest potency was exhibited by compound 64 with a Ki value of 0.23 μM.
In 2003, scientists from France and Greece reported the solution-phase synthesis of new phosphinopeptide inhibitors.94 Promising results were observed for RXP03, which was modified at the P1′ position by introducing an isoxazole group to obtain 65.
Research showed that compound 65 was 36-fold more active than RXP03 63 towards MMP-14. The investigation suggested that the P1′ substituent was responsible for the high potency and selectivity of these MMP inhibitors. Compound 65 was highly stereospecific; biological studies showed that the (R, S, S) diastereoisomer was 100-fold more active than the (R, R, S) isomer.
In 2005, Italian scientists reported the synthesis of three novel peptidomimetic phosphinate inhibitors and evaluated their biological activity towards metalloproteinases MMP-2 and MMP-8.95 All of the compounds were highly potent showing IC50 values in the micromolar range. The greatest activity was observed for inhibitors 66 against MMP-2.
The latest research reported the application of aminophosphinic acid derivatives for the treatment of Alzheimer's disease. They are classified as the inhibitors of β-secretase, an aspartic acid protease important in the formation of myelin sheaths in peripheral nerve cells.96 Until 2007, several pharmaceutical companies were in the early stages of testing new drugs for the treatment of Alzheimer's disease. In 2012, Merck reported the results of a phase I trial for MK-8931 67 and started a new clinical trial to evaluate the safety and effectiveness of this compound in patients with mild-to-moderate Alzheimer's disease. Phosphinic acid derivatives 68 as potential β-secretase inhibitors were first described in 2006.97 The authors of the patent reported that the synthesized inhibitors had high activity toward the enzyme with IC50 values in the range of 0.01–0.2 μM (Fig. 12).
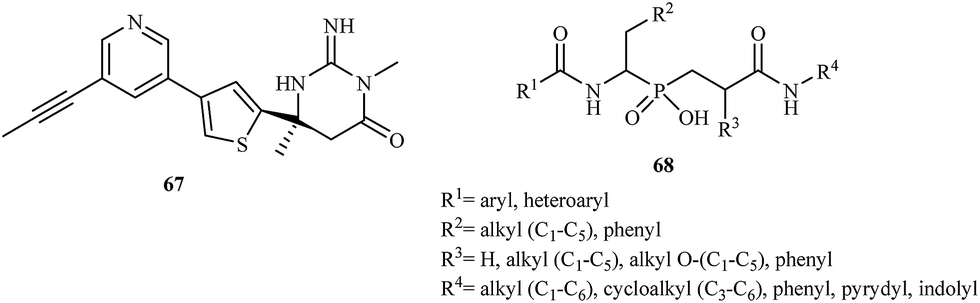 | ||
| Fig. 12 Structures of β-secretase inhibitors MK-8931 67 and 68. | ||
5.2. α-Aminophosphonic acid derivatives
α-Aminophosphonic acids derivatives are a significant type of compounds that contain a P–C bond. These class of molecules are known as effective chelating agents. The presence of a –NH2C–P(O)(OH)2 group increases their metal binding abilities. Many synthetic methods for the preparation of aminophosphonic acid derivatives have been described.98 Although aminophosphonic acids were mainly used as insecticides, herbicides, and plant-growth regulators, they have also been applied as effective chemotherapeutic agents.99A number of reports have been published on phosphonic acid derivatives used as MMP inhibitors. Hunter and coworkers reported the synthesis of a series of peptidomimetic α-aminophosphonic acid derivatives with high inhibitory properties against human fibroblast collagenase100 and carried out in vitro tests for these compounds. Introduction of a bromonaphthalimidoethyl group into the structure led to a very potent inhibitor 69 with an IC50 value of 0.02 μM. The (R, S, S) isomer of 69 was much more potent (80-fold) than the (S, S, S) isomer towards MMP-1.
In 1999, D'Alessio and coworkers synthesized and evaluated peptidomimetic N-(furan-2-yl)carbonyl-Leu-Trp-OH analogues.101 The compounds were tested for their activity against MMP-2, MMP-3, MMP-8, and MMP-9. The study showed that most peptidomimetic derivatives exhibited low potency towards MMPs. Nevertheless, compound 70 exhibited high activity with a satisfactory IC50 value of 60 μM.
Gallina et al. prepared a series of new phosphonic acid derivatives 71 by modifying the previously reported phosphotryptophan derivative L-Pro-L-Leu-L-(P)Trp(OH)2.102 By replacing the aminoterminal L-Pro with amino acid residues bearing small side chains, the affinity to MMP-2 and MMP-8 was increased, and the derivatives showed different selectivity profiles (Fig. 13).
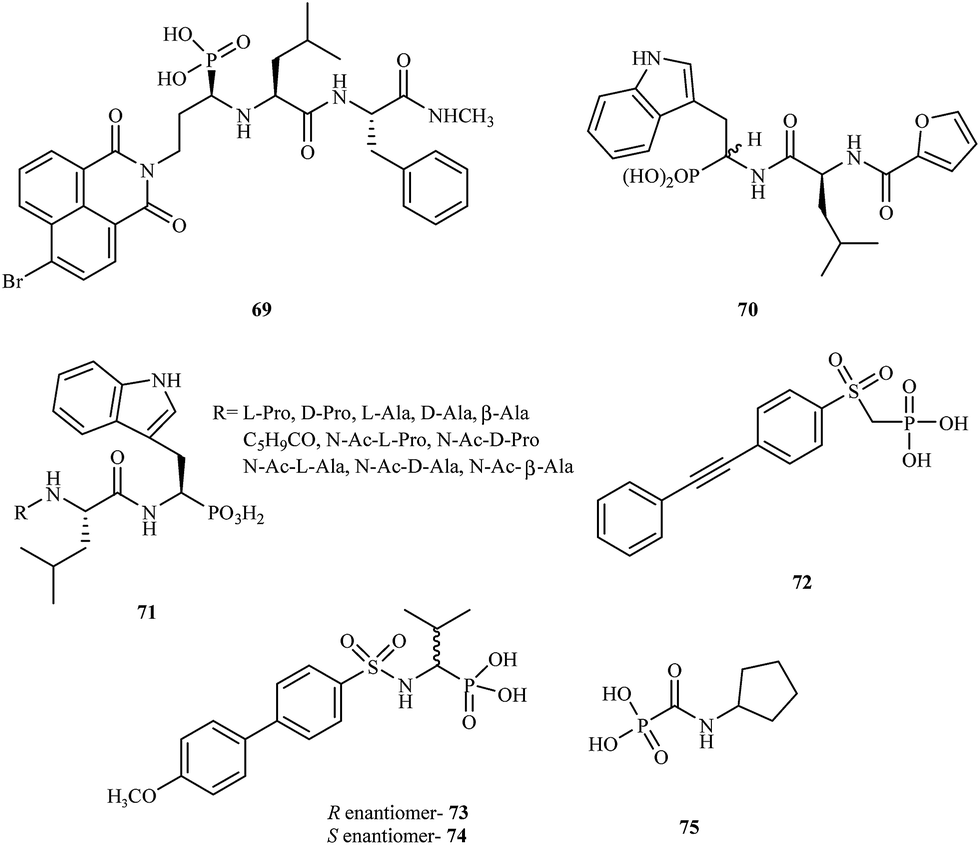 | ||
| Fig. 13 Chemical structures of aminophoshonic acid derivatives 69–75. | ||
In 2009, Tortorella and coworkers obtained new α-sulfonylaminophosphonate analogues.103 Preliminary activity screening of these compounds was investigated against MMP-2, MMP-8, MMP-13, and MMP-14. Most of the synthesized analogues were very effective MMP inhibitors, exhibiting IC50 values in the nanomolar range. Derivative 72 proved to be the most potent inhibitor towards MMP-2 with an IC50 = 60 nM.
The Mazza research group prepared new α-arylsulfonylamino phosphonates and tested them as stereoselective inhibitors of MMP-8.104 The mechanism of binding in the active site for both the R- and S-enantiomers (73 and 74, respectively) was explained by analyzing the crystal structures of the complexes with MMP-8. The study showed that enantiomer R 73 was much more potent than the S enantiomer 74 with an IC50 value in the nanomolar range.
Another class of phosphonic acid-based MMP inhibitors are the carbamoylphosphonic acid derivatives. A collaboration of scientists from Israel and Germany reported the synthesis, characterization and biological evaluation of the alkyl and cycloalkylcarbamoylphosphonic acid analogs.105 Their inhibition potency was tested in in vitro models with MMP-1, MMP-2, MMP-3, MMP-8, and MMP-9. The cycloalkylcarbamoylphosphonic acid derivatives exhibited higher potency compared to the open-chain alkyl analogs. Most of the analogs showed good selectivity against MMP-2. The optimal activity against MMP-2 was observed for compound 75 with an IC50 value of 0.08 μM. In 2005, the same research group reported106 that cycloalkylcarbamoylphosphonic acid-based inhibitors that block MMP-2 activity have a significant impact on the inhibition of tumor cell growth and reduce lung metastasis. This investigation proved that new carbamoyl phosphonate matrix metalloproteinase inhibitors might be effective drug candidates for cancer treatment.
In 2004, Breuer and coworkers prepared novel MMP inhibitors based on Ca(II), Mg(II), Zn(II) and Cu(II) complexes of cyclopentylcarbamoylphosphonic and 2-(N,N-dimethylamino)ethylcarbamoylphosphonic acids (76 and 77, respectively).107 Both carbamoyl phosphonates showed very high MMP-2 activity with IC50 values of 80 nM for compound 76 and 25 nM for analog 77. During the study, it was found that the dimethylamino group on compound 77 enhanced the binding potency of zinc binding group (ZBG) in aqueous solutions (Fig. 14).
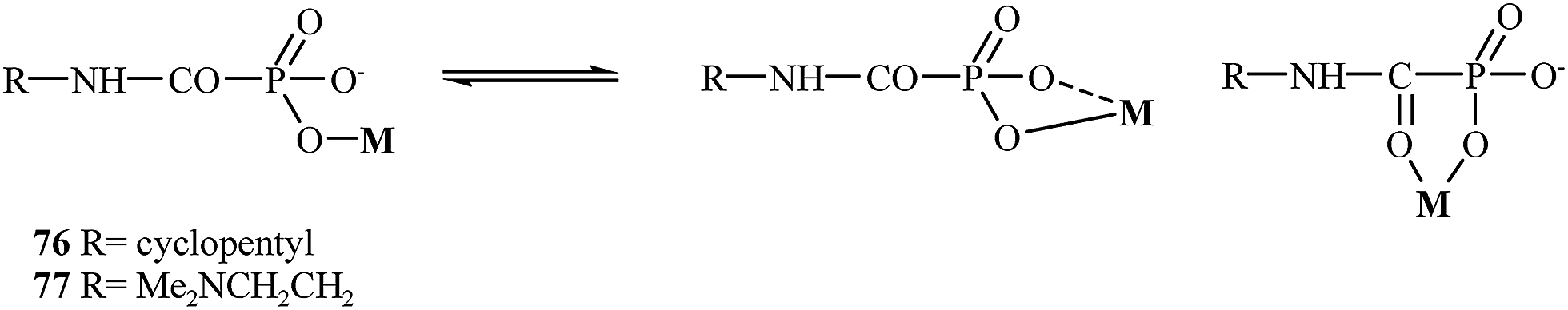 | ||
| Fig. 14 Structures of MMP inhibitors 76 and 77 based on metal complexes. | ||
In 2008, the Hoffman research group presented the synthesis of cis-2-aminocyclohexylcarbamoylphosphonic acid (cis-ACCP) 78 and analyzed its pharmacodynamic and pharmacokinetic properties.108 Cis-ACCP was evaluated in in vitro and in vivo cancer metastasis models and classified as a medium-potency MMP inhibitor; because of its good pharmacological profile, this molecule entered phase 2 and 3 clinical trials (Fig. 15).
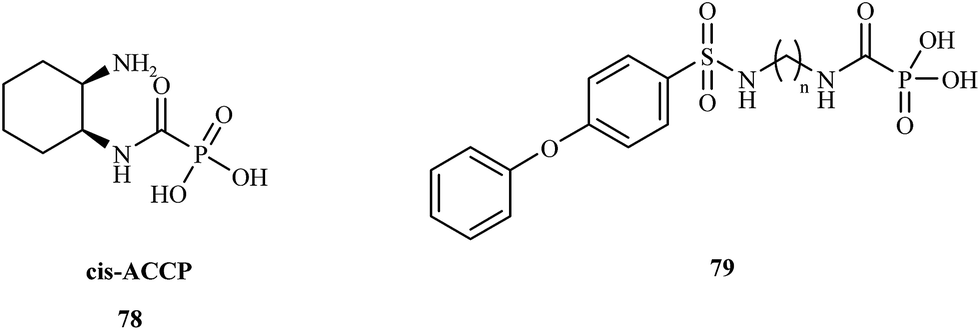 | ||
| Fig. 15 Structures of cis-ACCP 78 and MMP inhibitors 79. | ||
Recently, this laboratory109 presented the synthesis of seven 4-phenoxybenzenesulfonamidopolymethylene carbamoyl phosphonates 79 containing polymethylene chains. Biological evaluation of these compounds showed the highest potency for analogues with (CH2)5,6 groups, which exhibited antimetastatic activity in a murine melanoma model. Compounds with shorter polymethylene linkers were selective towards MMP-2 in contrast to the analogs bearing chains with 7 or 8 methylene groups, for which no inhibitor activity was observed.
The latest achievements in the development of aminophosphonic acids with biological activity are the synthesis and application of chiral thiourea derivatives. They are an important class of compounds that demonstrate potential antibacterial, fungicidal, antiviral (including HIV) and anticancer activity. Several of them have found practical application as herbicidal compounds or plant growth regulators.110 Currently, the use of chiral thiourea derivatives to treat disease is the subject of intense research.
6. Phosphorus compounds with antiviral activity
Development of new antiviral therapy is needed to treat viral infections that are not amenable to prophylaxis by vaccination or did not fulfill its promises for complete protection, but is also highly desirable for those infections where vaccination has not been implemented. The beginning of the antiviral era is marked by the description in 1959 of the synthesis of 5-iodo-2′-deoxyuridine (IDU).111 IDU was actually synthesized as a potential antitumor agent, but later became commercialized as the first antiviral drug to be used in the topical treatment of herpetic eye infections. Nowadays, among the drugs used in treatment of some viral infections [e.g. human immunodeficiency virus (HIV), hepatitis C virus (HCV), hepatitis B virus (HBV), cytomegalovirus (CMV), smallpox (variola virus)] there are many compounds containing a phosphorus atom.Nowadays, AIDS, caused by HIV infection is one of the major medical problems. According to the WHO data, 36.9 million people were living with HIV and 1.9 million people newly enrolled on antiretroviral treatment in 2014. In 1985, E. de Clercq et al., described the activity of 9-(R)-(2-phosphonomethoxypropyl)adenine (tenofovir) 80 against HIV in cell culture.112 Tenofovir is a nucleotide (nucleoside monophosphate) analogue with activity against retroviruses, including HIV-1, HIV-2 and hepadnaviruses in a variety of cell types, including resting cells. Tenofovir is administered to patients in the form of a prodrug – tenofovir disoproxil fumarate (tenofovir DF) 81. Following absorption, tenofovir DF is rapidly converted to tenofovir (Fig. 16), which is metabolised intracellularly to its active anabolite, which is a competitive inhibitor of HIV-1 reverse transcriptase and terminates the growing DNA chain.113
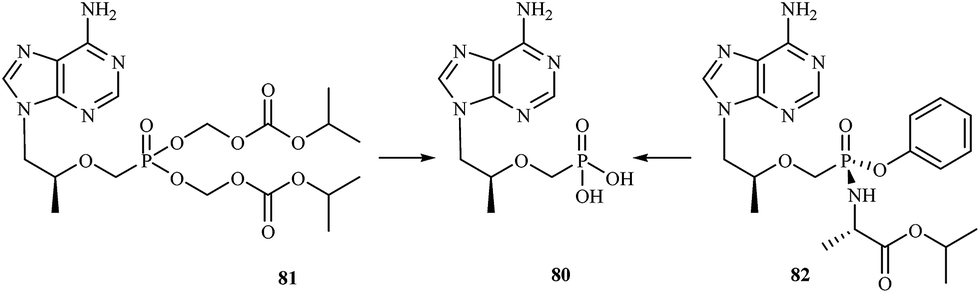 | ||
| Fig. 16 Conversion of tenofovir DF 81 and GS-7340 82 to tenofovir 80. | ||
Continuing research into more active agents led to the discovery of other derivatives of tenofovir with better distribution into lymphoid tissues. GS-7340 82 is a prototype molecule representing a novel class of tenofovir mono-phosphonoamidate prodrugs. Unlike tenofovir, GS-7340 contains phenol and alanine isopropyl ester as the phosphonate masking groups. Relative to parent tenofovir, GS-7340 exhibits 500- to 1000-fold enhanced activity against HIV-1 in T-cells, activated peripheral blood mononuclear lymphocytes and macrophages.114
There are currently a large number of agents in development across a variety of classes for the treatment of HCV infections. One class for which promising in vitro results have been reported is represented by the nucleoside/nucleotide analogs. These compounds share properties with the intracellular nucleoside substrates of the target HCV enzymes involved in the transcription of the viral genome and, when phosphorylated to the nucleoside-triphosphate, lead to premature termination of the growing HCV RNA chain during viral replication.115 One of nucleotide analog, sofosbuvir 83 is a phosphoramidate prodrug that is metabolized within the liver into the active antiviral agent 2′-deoxy-2′-α-fluoro-β-C-methyluridine-5′-monophosphate 84 which is further phosphorylated to the active 2′-deoxy-2′-α-fluoro-β-C-methyluridine-5′-triphosphate 85 (Fig. 17).116 The triphosphate serves as a defective substrate for the NS5B protein, which is the viral RNA polymerase, thus acts as an inhibitor of viral RNA synthesis.117 In 2013, the Food and Drug Administration (FDA) approved sofosbuvir in combination with ribavirin for oral dual therapy of HCV genotypes 2 and 3, and for triple therapy with injected pegylated interferon and ribavirin for treatment-naive patients with HCV genotypes 1 and 4. In 2014 a combination of sofosbuvir with the viral NS5A inhibitor ledipasvir was approved.
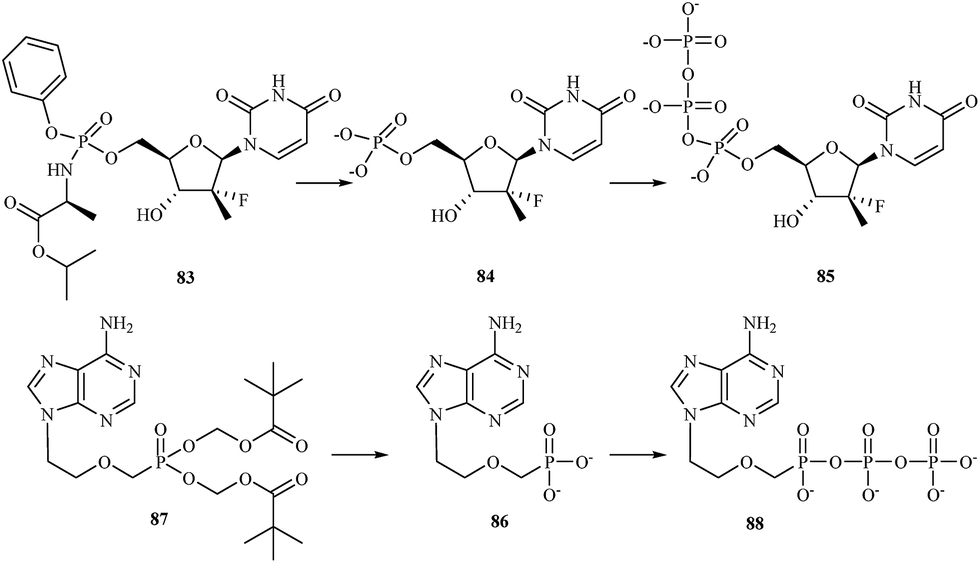 | ||
| Fig. 17 Activation mechanism of sofosbuvir 83 and adefovir 86. | ||
Hepatitis B is another viral infection that attacks the liver and can cause both acute and chronic disease. According to the WHO data, an estimated 240 million people worldwide are chronically infected with HBV. More than 780000 people die every year due to complications of hepatitis B, including cirrhosis or liver cancer.118 Adefovir 86 is a drug used to treat infections with hepatitis B virus. It is a nucleotide analog with reverse transcriptase inhibitory activity. Adefovir is orally administrated as a prodrug – adefovir dipivoxil 87. After administration adefovir dipivoxil is hydrolysed to adefovir and phosphorylated to its active diphosphorylated form adefovir dipivoxil 88 (Fig. 17). Upon phosphorylation, adefovir DP competes with dATP for incorporation by the HBV reverse transcriptase. The lack of a 3′-hydroxyl group causes chain-termination when adefovir is incorporated into viral transcripts.119 Dose of 10 mg per day of adefovir dipivoxil significantly improved histological, biochemical and virological outcomes in HBeAg-positive and – negative patients, and serological outcomes in HBeAg-positive patients.120
Treatment of CMV infections in immunosuppressed patients based upon several compounds including foscarnet 89 and cidofovir 90. Both compounds are administered intravenously [foscarnet at 180 mg kg−1 per day for induction therapy and at 120 mg kg−1 per day for maintenance therapy; cidofovir at 5 mg kg−1 per week during the first 2 weeks (induction therapy), and then 5 mg kg−1 every other week (maintenance therapy)]. Foscarnet and cidofovir are targeted at the viral DNA polymerase. Foscarnet interacts directly with the pyrophosphate binding site of the DNA polymerase, whereas cidofovir must be first phosphorylated to its diphosphate derivative, which then interact as competitive inhibitor/alternate substrates. As alternate substrate phosphorylated cidofovir is incorporated into the growing DNA chain and block chain elongation (Fig. 18).121
 | ||
| Fig. 18 Structures of foscarnet 89, cidofovir 90 and CMX-001 91. | ||
With the declaration by WHO in 1980 that smallpox had been eradicated from the earth, any attempts to develop a potentially active anti-poxvirus drug were abandoned. In 2001, the fear that variola virus might emerge again as the consequence of a terrorist attack. In the US a program was launched to identify antiviral agents that could be used prophylactically or therapeutically against orthopoxviruses, which could be employed as a biological weapon or to give arise to an inadvertent outbreak. One of the foremost candidate to be used in such scenario is CMX-001 91. CMX-001 is the hexadecyloxypropyl ester of cidofovir, which had already been reported as an antiviral agent active against vaccinia virus. CMX-001 is a highly promising compounds to treat pathogenic orthopoxvirus infection in humans and should not only be intended for therapeutic use against smallpox, but also for progressive vaccinia as the consequence of the smallpox vaccination (in immunosuppressed patients); monkeypox, where cidofovir has proved more efficacious than vaccination; and even molluscum contagiosum (due to molluscipoxvirus).4
7. Conclusions
This literature review has emphasized and described the importance of organophosphorus derivatives, which are a wide class of chemical compounds containing organic moieties usually bonded directly to phosphorus or bonded through a heteroatom, such as sulfur, oxygen or nitrogen. As discussed in this literature review, medical applications of organophosphorus compounds as drugs or drug candidates against many diseases have expanded greatly in recent years. OPs are used in clinical practice as compounds with anticancer or antiviral properties. Bisphosphonates are currently the most important and effective class of drugs developed for the treatment of metabolic bone disorders associated with increased osteoclast-mediated bone resorption, such as osteoporosis. Aminophosphinic and aminophosphonic acids also are a very important group of chemical compounds, and their synthesis has attracted considerable attention in medicinal chemistry. They are classified as antimetabolites, which compete with their aminocarboxylic acid analogues in the active sites of enzymes and other cell receptors. It is without a doubt because of the unique properties and various applications of organophosphorus compounds that will continue to make them the subject of intense research investigations around the world.List of abbreviations
| ACE | Angiotensin converting enzyme |
| CA4P | Combretastatin A-4 phosphate |
| CMV | Cytomegalovirus |
| CWA | Chemical warfare agents |
| FDA | Food and drug administration |
| HBV | Hepatitis B virus |
| HCV | Hepatitis C virus |
| HIV | Human immunodeficiency virus |
| IDU | 5-Iodo-2′-deoxyuridine |
| MMPs | Matrix metalloproteinases |
| OPs | Organophosphorus compounds |
| PAN | Pesticide action network |
| SERMs | Specific estrogen receptor modulators |
| STS | Steroid sulfatase |
| TEPP | Tetraethyl pyrophosphate |
| thioTEPA | N,N′,N′′-Triethylenethiophosphoramide |
| WHO | World health organization |
| ZBG | Zinc binding group |
Acknowledgements
We gratefully acknowledge the National Science Centre (Poland) for financial support (grant no. 2011/03/D/NZ7/03985).References
- B. K. Singh and A. Walker, FEMS Microbiol. Rev., 2006, 30, 428–471 CrossRef CAS PubMed.
- A. Marklund, B. Andersson and P. Haglund, Chemosphere, 2003, 53, 1137–1146 CrossRef CAS PubMed.
- H. R. Hudson, N. J. Wardle, S. W. Bligh, I. Greiner, A. Grun and G. Keglevich, Mini-Rev. Med. Chem., 2012, 12, 313–325 CrossRef CAS PubMed.
- E. de Clercq, Biochem. Pharmacol., 2013, 85, 727–744 CrossRef CAS PubMed.
- Q. A. McKellar and F. Jackson, Trends Parasitol., 2004, 20, 456–461 CrossRef CAS PubMed.
- T. C. Marrs, R. L. Maynard and F. R. Sidell, Chemical Warfare Agents: Toxicology and Treatment, John Wiley & Sons Ltd, Chinchester, 2nd edn, 2007 Search PubMed.
- A. V. Terry Jr, Pharmacol. Ther., 2012, 134, 355–365 CrossRef CAS PubMed.
- European Commission, Report on osteoporosis in the European Community- Action for prevention, 1998 Search PubMed.
- P. D. Delmas, Lancet, 2002, 359, 2018–2026 CrossRef CAS.
- J. A. Kanis, N. Burlet, C. Cooper, P. D. Delmas, J. Y. Reginster, F. Borgstrom and R. Rizzoli, Osteoporosis Int., 2008, 19, 399–428 CrossRef CAS PubMed.
- P. D. Delmas and P. J. Meunier, N. Engl. J. Med., 1997, 336, 558–566 CrossRef CAS PubMed.
- C. Roux and M. Dougados, Drugs, 1999, 58, 823–830 CrossRef CAS PubMed.
- R. E. Coleman, Oncologist, 2004, 9, 14–27 CrossRef CAS PubMed.
- H. Fleisch, Bisphosphonates in Bone Disease. From the Laboratory to the Patient, Academic Press, San Diego, 2000 Search PubMed.
- P. Clezardin, Bone, 2011, 48, 71–79 CrossRef CAS PubMed.
- H. Fleisch, The Aging Spine, 2005, 2, 60–64 Search PubMed.
- H. Fleisch, R. G. G. Russell, S. Bisaz, P. A. Casey and R. C. Mühlbauer, Calcif. Tissue Res., 1968, 2, 10 CrossRef.
- M. D. Francis, R. G. G. Russell and H. Fleisch, Science, 1969, 165, 1264–1266 CAS.
- H. Fleisch, R. G. G. Russell and M. D. Francis, Science, 1969, 165, 1262–1264 CAS.
- H. Fleisch, R. G. G. Russell, B. Simpson and R. C. Mühlbauer, Nature, 1969, 223, 211–212 CrossRef CAS PubMed.
- J. M. van Gelder, E. Breuer, A. Ornoy, A. Schlossman, N. Patlas and G. Golomb, Bone, 1995, 16, 511–520 CrossRef CAS PubMed.
- J. M. van Gelder, E. Breuer, A. Schlossman, A. Ornoy, J. Monkkonen, J. Simila, T. Klenner, H. Stadler, B. Krempien, N. Patlas and G. Golomb, J. Pharm. Sci., 1997, 86, 283–289 CrossRef PubMed.
- H. Cohen, V. Solomon, I. S. Alferiev, E. Breuer, A. Ornoy, N. Patlas, N. Eidelman, G. Hagele and G. Golomb, Pharm. Res., 1998, 15, 606–613 CrossRef CAS.
- E. Breuer, H. Zaher, Z. Tashma and D. Gibson, Heteroat. Chem., 1996, 7, 515–520 CrossRef CAS.
- J. R. Ross, Y. Saunders, P. M. Edmonds, S. Patel, D. Wonderling, C. Normand and K. Broadley, Health Tech. Asses., 2004, 8, 1–176 CAS.
- S. R. Fletcher, J. J. Kulagowski and M. R. Teall, Eur. Pat., 485026, 1992.
- C. P. Ciosek Jr, D. R. Magnin, T. W. Harrity, J. V. H. Logan, J. K. Dickson Jr, E. M. Gordon, K. A. Hamilton, K. G. Jolibois and L. K. Kunselman, J. Biol. Chem., 1993, 268, 24832–24837 CAS.
- M. J. van Maanen, C. J. M. Smeets and J. H. Beijnen, Cancer Treat. Rev., 2000, 26, 257–268 CrossRef CAS PubMed.
- D. J. Taylor, C. E. Parsons, H. Han, A. Jayaraman and K. Rege, BMC Cancer, 2011, 11, 470 CrossRef CAS PubMed.
- S. McCracken and J. Wolf, Cancer Chemother. Rep., 1960, 6, 52–54 Search PubMed.
- V. A. Chernov, A. A. Grushina and L. G. Lytkina, Farmakol Toksikol, 1963, 26, 102–108 CAS.
- C. W. Noell and C. C. Cheng, J. Med. Chem., 1968, 11, 63–66 CrossRef CAS PubMed.
- G. Sosnovsky, N. O. Maheswara and S. W. Li, J. Med. Chem., 1986, 29, 2225–2230 CrossRef CAS PubMed.
- F. Baumann and R. Preiss, J. Chromatogr. B: Biomed. Sci. Appl., 2001, 764, 173–192 CrossRef CAS.
- T. Wagner, Clin. Pharmacokinet., 1994, 26, 439–456 CrossRef CAS PubMed.
- C. Joqueviel, V. Gilard, R. Martino, M. Malet-Martino and U. Niemeyer, Cancer Chemother. Pharmacol., 1997, 40, 391–399 CrossRef CAS PubMed.
- S. B. Murphy, W. P. Bowman, M. Abromowitch, J. Mirro, J. Ochs, G. Rivera, C. H. Pui, D. Fairclough and C. W. Berard, J. Clin. Oncol., 1986, 4, 1732–1739 CAS.
- C. Flader, J. Liu and F. Borch, J. Med. Chem., 2000, 43, 3157–3167 CrossRef CAS PubMed.
- J. X. Duan, H. Jiao, J. Kaizerman, T. Stanton, J. W. Evans, L. Lan, G. Lorente, M. Banica, D. Jung, J. Wang, H. Ma, X. Li, Z. Yang, R. M. Hoffman, W. S. Ammons, C. P. Hart and M. Matteucciat, J. Med. Chem., 2008, 51, 2412–2420 CrossRef CAS PubMed.
- R. F. Borch, J. Liu, J. P. Schmidt, J. T. Marakovits, C. Joswig, J. J. Gipp and R. T. Mulcahy, J. Med. Chem., 2000, 43, 2258–2265 CrossRef CAS PubMed.
- M. Jain and C. H. Kwon, J. Med. Chem., 2003, 46, 5428–5436 CrossRef CAS PubMed.
- C. J. Anderson, L. J. H. Lucas and T. S. Widlanski, J. Am. Chem. Soc., 1995, 17, 3889–3890 CrossRef.
- W. Kozak, M. Daśko, M. Masłyk, J. S. Pieczykolan, B. Gielniewski, J. Rachon and S. Demkowicz, RSC Adv., 2014, 4, 44350–44358 RSC.
- W. Kozak, M. Daśko, M. Masłyk, B. Gielniewski, J. Rachon and S. Demkowicz, J. Asian Nat. Prod. Res., 2015, 17, 1091–1096 CrossRef CAS PubMed.
- W. Kozak, M. Daśko, A. Wołos, M. Masłyk, K. Kubiński, A. Składanowski, M. Misiak, J. Rachon and S. Demkowicz, RSC Adv., 2015, 5, 32594–32603 RSC.
- S. Demkowicz, W. Kozak, M. Daśko, M. Masłyk, K. Kubiński and J. Rachon, Drug Dev. Res., 2015, 76, 94–104 CrossRef CAS PubMed.
- W. Kozak, M. Daśko, M. Masłyk, K. Kubiński, J. Rachon and S. Demkowicz, Drug Dev. Res., 2015, 76, 450–462 CrossRef CAS PubMed.
- S. Demkowicz, W. Kozak, M. Daśko, M. Masłyk, B. Gielniewski and J. Rachon, Eur. J. Med. Chem., 2015, 101, 358–366 CrossRef CAS PubMed.
- S. A. David, A. V. Hallum III, M. Stratton-Custis, D. J. Garcia, M. Gleason-Guzman, S. E. Salmon, P. Santabarbara, E. J. Niesor, S. Floret and C. L. Bentzen, Clin. Cancer Res., 2001, 7, 1246–1250 Search PubMed.
- P. J. Kuehl, S. P. Stratton, M. B. Powell and P. B. Myrdal, Int. J. Pharm., 2009, 382, 104–110 CrossRef CAS PubMed.
- A. Dowlati, K. Robertson, M. Cooney, W. P. Petros, M. Stratford, J. Jesberger, N. Rafie, B. Overmoyer, V. Makkar, B. Stambler, A. Taylor, J. Waas, J. S. Lewin, K. R. McCrae and S. C. Remick, Cancer Res., 2002, 62, 3408–3416 CAS.
- G. J. Russell and E. Lacey, Biochem. Mol. Biol. Int., 1995, 35, 1153–1159 CAS.
- B. Nawrot, B. Rebowska, O. Michalak, M. Bulkowski, D. Blaziak, P. Guga and W. J. Stec, Pure Appl. Chem., 2008, 80, 1859–1871 CrossRef CAS.
- R. Z. Yu, J. S. Grundy and R. S. Geary, Expert Opin. Drug Metab. Toxicol., 2013, 9, 169–182 CrossRef CAS PubMed.
- T. Sugiyama and A. Kittaka, Molecules, 2013, 18, 287–310 CrossRef CAS PubMed.
- P. C. Zamecnik and M. L. Stephenson, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 280–284 CrossRef CAS.
- B. Rosenberg, L. van Camp and T. Krigas, Nature, 1965, 205, 698–699 CrossRef CAS PubMed.
- I. Kostova, Anti-Cancer Agents Med. Chem., 2006, 6, 19–32 CrossRef CAS PubMed.
- J. Reedijk and E. Buowman, Bioinorganic Catalysis, Dekker, New York, 1999 Search PubMed.
- S. J. Lippard, Science, 1993, 261, 699–700 CAS.
- M. J. Clarke, F. Zhu and D. R. Frasca, Chem. Rev., 1999, 99, 2511–2534 CrossRef CAS PubMed.
- A. Bergamo, C. Gaiddon, J. H. Schellens, J. H. Beijnen and G. Sava, J. Inorg. Biochem., 2012, 106, 90–99 CrossRef CAS PubMed.
- F. Lentz, A. Drescher, A. Lindauer, M. Henke, R. A. Hilger, C. G. Hartinger, M. E. Scheulen, C. Dittrich, B. K. Keppler and U. Jaehde, Anticancer Drugs, 2009, 20, 97–103 CrossRef CAS PubMed.
- C. Scolaro, A. Bergamo, L. Brescacin, R. Delfino, M. Cocchietto, G. Laurenczy, T. J. Geldbach, G. Sava and P. J. Dyson, J. Med. Chem., 2005, 48, 4161–4171 CrossRef CAS PubMed.
- S. Komeda and A. Casini, Curr. Top. Med. Chem., 2012, 12, 219–235 CrossRef CAS PubMed.
- W. H. Ang and P. J. Dyson, Eur. J. Inorg. Chem., 2006, 20, 4003–4018 CrossRef.
- K. Chakree, C. Ovatlarnporn, P. J. Dyson and A. Ratanaphan, Int. J. Mol. Sci., 2012, 13, 13183–13202 CrossRef CAS PubMed.
- A. A. Nazarov, S. M. Meier, O. Zava, Y. N. Nosova, E. R. Milaeva, C. G. Hartinger and P. J. Dyson, Dalton Trans., 2015, 44, 3614–3623 RSC.
- W. S. Knowels, M. J. Sabacky and D. B. Vineyard, Adv. Chem. Ser., 1974, 132, 274–282 CrossRef.
- J. P. Roberts and C. Lee, Org. Lett., 2005, 7, 2679–2682 CrossRef CAS PubMed.
- N. Jeong, B. K. Sung and Y. K. Choi, J. Am. Chem. Soc., 2000, 122, 6771–6772 CrossRef CAS.
- Y. Wei and M. Shi, Chem. Res. Toxicol., 2010, 43, 1005–1018 CAS.
- T. Shibata, K. Takasaku, Y. Takesue, N. Hirata and K. Takagi, Synlett, 2002, 10, 1681–1682 CrossRef.
- S. L. Shi, L. W. Xu, K. Oisaki, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2010, 132, 6638–6639 CrossRef CAS PubMed.
- W. S. Knowles, M. J. Sabacky and D. B. Vineyard, J. Chem. Soc., Chem. Commun., 1972, 1, 10–11 RSC.
- A. Concepción, M. González, M. Fuertes, G. Rubiales, J. M. Ezpeleta and F. Palacios, J. Org. Chem., 2013, 78, 3858–3866 CrossRef PubMed.
- A. Mucha, P. Kafarski and L. Berlicki, J. Med. Chem., 2011, 54, 5955–5980 CrossRef CAS PubMed.
- F. Palacios, C. Alonso and J. M. de Los Santos, Chem. Rev., 2005, 105, 899–931 CrossRef CAS PubMed.
- P. Kafarski and B. Lejczak, Curr. Med. Chem.: Anti-Cancer Agents, 2001, 1, 301–312 CrossRef CAS PubMed.
- A. Yiotakis, A. Lecoq, S. Vassiliou, I. Raynal, P. Cuniasse and V. Dive, J. Med. Chem., 1994, 37, 2713–2720 CrossRef CAS PubMed.
- P. A. Bartlett and W. B. Kezer, J. Am. Chem. Soc., 1984, 106, 4282–4283 CrossRef CAS.
- B. Vincent, J. Jiracek, F. Noble, M. Loog, B. Roques, V. Dive, J. P. Vincent and F. Checler, Br. J. Pharmacol., 1997, 121, 705–710 CrossRef CAS PubMed.
- P. van Lint and C. Libert, J. Leukocyte Biol., 2007, 82, 1375–1381 CrossRef CAS PubMed.
- J. A. Jacobsen, J. L. M. Jourden, M. T. Miller and S. M. Cohen, Biochim. Biophys. Acta, Mol. Cell Res., 2010, 1803, 72–94 CrossRef CAS PubMed.
- P. Vihinen and V. M. Kähäri, Int. J. Cancer, 2002, 99, 157–166 CrossRef CAS PubMed.
- J. P. Witty, S. A. Foster, G. P. Stricklin, L. M. Matrisian and P. H. Stern, J. Bone Miner. Res., 1996, 11, 72–78 CrossRef CAS PubMed.
- D. Ahrens, A. E. Koch, R. M. Pope, M. Steinpicarella and M. J. Niedbala, Arthritis Rheum., 1996, 39, 1576–1587 CrossRef CAS PubMed.
- D. C. Calentano and W. H. Frishman, J. Clin. Pharmacol., 1997, 37, 991–1000 CrossRef.
- J. F. Woessner Jr, Ann. N. Y. Acad. Sci., 1994, 732, 11–21 CrossRef CAS.
- M. Ebata, Y. Fukuda, I. Nakano, Y. Katano, N. Fujimoto and T. Hayakawa, Liver, 1997, 17, 293–299 CrossRef CAS PubMed.
- J. Krapcho, C. Turk, D. W. Cushman, J. R. Powell, J. M. de Forrest, E. R. Spitzmiller, D. S. Karanewsky, M. Duggan, G. Rovnsak, J. Schwartz, S. Natarajan, J. D. Godfrey, D. E. Ryono, R. Neubeck, K. S. Atwa and E. W. Petrillo Jr, J. Med. Chem., 1988, 31, 1148–1160 CrossRef CAS PubMed.
- S. Vassiliou, A. Mucha, P. Cuniasse, D. Georgiadis, K. Lucet-Levannier, F. Beau, R. Kannan, G. Murphy, V. Knäuper, M. C. Rio, P. Basset, A. Yiotakis and V. Dive, J. Med. Chem., 1999, 42, 2610–2620 CrossRef CAS PubMed.
- M. Matziari, F. Beau, P. Cuniasse, V. Dive and A. Yiotakis, J. Med. Chem., 2004, 47, 325–336 CrossRef CAS PubMed.
- A. Makaritis, D. Georgiadis, V. Dive and A. Yiotakis, Chem.–Eur. J., 2003, 9, 2079–2094 CrossRef CAS PubMed.
- G. Bianchini, M. Aschi, G. Cavicchio, M. Crucianelli, S. Preziuso, C. Gallina, A. Nastari, E. Gavuzzo and F. Mazza, Bioorg. Med. Chem., 2005, 13, 4740–4749 CrossRef CAS PubMed.
- M. Willem, A. N. Garratt, B. Novak, M. Citron, S. Kaufmann, A. Rittger, B. de Strooper, P. Saftig, C. Birchmeier and C. Haass, Science, 2006, 314, 664–666 CrossRef CAS PubMed.
- H. Hilpert, R. Humm, D. Knopp and P. Weiss, Eur. Pat., 1685142A1, 2006.
- E. D. Naydenova, P. T. Todorov and K. D. Troev, Amino Acids, 2010, 38, 23–30 CrossRef CAS PubMed.
- J. B. Camden, US Pat., 6090796, 2000.
- D. J. Hunter, J. Bird, F. Cassidy, R. C. de Mello, G. P. Harper, E. H. Karran, R. E. Markwell, A. J. Miles-Williams and R. W. Ward, Bioorg. Med. Chem. Lett., 1994, 4, 1833–1836 CrossRef CAS.
- S. D'Alessio, C. Gallina, E. Gavuzzo, C. Giordano, B. Gorini, F. Mazza, M. Paglialunga Paradisi, G. Panini, G. Pochetti and A. Sella, Bioorg. Med. Chem., 1999, 7, 389–394 CrossRef.
- M. Agamennone, C. Campestre, S. Preziuso, V. Consalvi, M. Crucianelli, F. Mazza, V. Politi, R. Ragno, P. Tortorella and C. Gallina, Eur. J. Med. Chem., 2005, 40, 271–279 CrossRef CAS PubMed.
- M. T. Rubino, M. Agamennone, C. Campestre, G. Fracchiolla, A. Laghezza, F. Loiodice, E. Nuti, A. Rossello and R. Tortorella, ChemMedChem, 2009, 4, 352–362 CrossRef CAS PubMed.
- G. Pochetti, E. Gavuzzo, C. Campestre, M. Agamennone, P. Tortorella, V. Consalvi, C. Gallina, O. Hiller, H. Tschesche, P. A. Tucker and F. Mazza, J. Med. Chem., 2006, 49, 923–931 CrossRef CAS PubMed.
- E. Breuer, C. J. Salomon, Y. Katz, W. Chen, S. Lu, G. V. Röschenthaler, R. Hadar and R. Reich, J. Med. Chem., 2004, 47, 2826–2832 CrossRef CAS PubMed.
- R. Reich, Y. Katz, R. Hadar and E. Breuer, Clin. Cancer Res., 2005, 11, 3925–3929 CrossRef CAS PubMed.
- E. Farkas, Y. Katz, S. Bhusare, R. Reich, G. V. Röschenthaler, M. Königsmann and E. Breuer, JBIC, J. Biol. Inorg. Chem., 2004, 9, 307–315 CrossRef CAS PubMed.
- A. Hoffman, B. Quadri, J. Frant, Y. Katz, S. R. Bhusare, E. Breuer, R. Hadar and R. Reich, J. Med. Chem., 2008, 51, 1406–1414 CrossRef CAS PubMed.
- J. Frant, A. Veerendhar, T. Chernilovsky, S. Nedvetzki, O. Vaksman, A. Hoffman, E. Breuer and R. Reich, ChemMedChem, 2011, 6, 1471–1477 CrossRef CAS PubMed.
- J. Liu, S. Yang, X. Li, H. Fan, P. Bhadury, W. Xu, J. Wu and Z. Wang, Molecules, 2010, 15, 5112–5123 CrossRef CAS PubMed.
- W. H. Prusoff, Biochim. Biophys. Acta, 1959, 32, 295–296 CrossRef CAS.
- E. de Clercq, A. Holy and I. Rosenberg, US Pat., 4724233A, 1985.
- B. P. Kearney, J. F. Flaherty and J. Shah, Clin. Pharmacokinet., 2004, 43, 595–612 CrossRef CAS PubMed.
- W. A. Lee, H. Gong-Xin, E. Eisenberg, T. Cihlar, S. Swaminathan, A. Mulato and K. Cundy, Antimicrob. Agents Chemother., 2005, 49, 1898–1906 CrossRef CAS PubMed.
- A. B. Eldrup, M. Prhavc, J. Brooks, B. Bhat, T. P. Prakash, Q. Song, S. Bera, N. Bhat, P. Dande, P. Dan Cook, C. F. Bennett, S. S. Carroll, R. G. Ball, M. Bossserman, C. Burlein, L. F. Colwell, J. F. Fay, O. A. Flores, K. Getty, R. L. LaFemina, J. Leone, M. MacCoss, D. R. McMasters, J. E. Tomassini, D. von Langen, B. Wolanski and D. B. Olsen, J. Med. Chem., 2004, 47, 5284–5297 CrossRef CAS PubMed.
- M. J. Sofia, D. Bao, W. Chang, J. Du, D. Nagarathnam, S. Rachakonda, P. G. Reddy, B. S. Ross, P. Wang, H. R. Zhang, S. Bansal, C. Espiritu, M. Keilman, A. M. Lam, H. M. Micolochick Steuer, C. Niu, M. J. Otto and P. A. Furman, J. Med. Chem., 2010, 53, 7202–7218 CrossRef CAS PubMed.
- A. Fung, Z. Jin, N. Dyatkina, G. Wang, L. Beigelman and J. Deval, Antimicrob. Agents Chemother., 2014, 58, 3636–3645 CrossRef PubMed.
- World Health Organization, Hepatitis B. Fact sheet N°204, July 2015, http://www.who.int/mediacentre/factsheets/fs204/en/, Accessed September 21, 2015 Search PubMed.
- A. S. Ray, J. E. Vela, L. Olson and A. Fridland, Biochem. Pharmacol., 2004, 68, 1825–1831 CrossRef CAS PubMed.
- T. M. Dando and G. L. Plosker, Drugs, 2003, 63, 2215–2234 CrossRef CAS PubMed.
- E. de Clercq, J. Antimicrob. Chemother., 2003, 51, 1079–1083 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |