
Phase evolution and crystal growth of VO2 nanostructures under hydrothermal reactions
Abstract
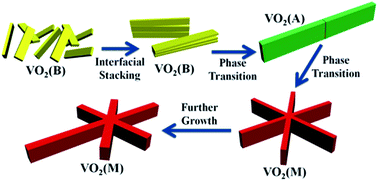
The phase evolution and crystal growth of VO2 nanostructures against reaction time in a high-pressure V2O5–oxalic acid hydrothermal system were systematically investigated. It was found that the rather thin VO2 (B) nanobelts were first obtained, then stacked to form large belt-like structures and subsequently phase transformed into VO2 (A), based on an oriented attachment-recrystallization mechanism. The large VO2 (A) belt-like structures could further assemble into novel “snowflake” VO2 (M) microcrystals with even bigger sizes and nearly well-defined six-fold symmetry. Due to the Ostwald ripening effect regarding crystal size discrepancy, the VO2 (M) phase could further grow at the cost of the gradual dissolution of VO2 (A) and full elimination of VO2 (B). The phase evolution from VO2 (B) first to VO2 (A) and then to VO2 (M), is actually a step-by-step thermodynamically downhill process, owing to the gradual relaxation of structural tension within the VO2 crystal lattice. Thus, our investigation, for the first time, demonstrated the feasibility of the well-known Ostwald's step rules towards the phase evolution process of VO2 and could provide unprecedented new insight to promote understanding of the synthesis and properties of vanadium oxide compounds.
 Please wait while we load your content...
Please wait while we load your content...

