
Investigation of proton-driven amine functionalized tube array as ion responsive biomimetic nanochannels†
Qiao-Ling Ma‡
,
Hong Xia‡,
Shou-Ting Zhang,
Dong-Dong Qin,
Samrat Devaramani,
Duo-Liang Shan and
Xiao-Quan Lu*
Key Laboratory of Bioelectrochemistry & Environmental Analysis of Gansu Province, College of Chemistry & Chemical Engineering, Northwest Normal University, Lanzhou, 730070, China. E-mail: luxq@nwnu.edu.cn; Fax: +86-931-7971323; Tel: +86-931-7971276
First published on 28th December 2015
Abstract
A simple amine embellished tube array was assembled at the liquid–liquid interface to study ion transfer behavior. Variation in the pH of the solution resulted in three different protonation states at the amino groups of the nanochannel, which in turn regulated ion transport, similar to the switching effect of ion channels in vivo.
Introduction
The living cells that are undergoing metabolism ceaselessly must continue to exchange material with the surrounding environment. There exists various nanochannels in the cell membranes of organisms, which play an important role in cellular energy conversion, signal transmission and functional regulation of molecular biological process.1–4 Hodgkin and Huxley described cellular currents by the concerted action of voltage-gated channels and first put forward the concept of ion channels in 1952.5 Neher and Sakman invented the patch-clamping technique, which revolutionized the electrophysiological investigation of ion channels.6–9 Many methods, such as molecular dynamics simulations,10 spectroscopy,11 biochemical and electrophysiological methods,12 have been extensively used to probe ion channels. For the past few years, the bio-inspired study towards the design and development of biomimetic nanochannels to simulate biological processes has attracted a great deal of attention.13 Biomimetic nanochannels, such as anion-doped carbon nanotubes,14 supramolecular channels15 and track-etched polymer membranes,16 have emerged as possible candidates for imitating physiological processes found in organisms.Nanochannels have been well studied for the purpose of building smart materials for various applications. Currently, nanochannels were prepared from the materials of various origins such as biological, organic and inorganic. When compared to biological, these organic and inorganic ion channels17 have better stability and greater flexibility in terms of shape and size. Above all, the nanochannels expose surface active chemical groups, which provide a good foundation for functionalization. Porous anodic alumina (AAO) films offer good stability and easy tailoring with uniform nanotube arrays, which make them ideal to construct ion channels and used to investigate cross-channel ion transfer.18 In general, ion transfer in living organisms is very complex and is restricted in a certain space. Therefore, it is absolutely essential to construct biomimetic nanochannels that can provide a simple and efficient method to reveal the complex life process. A liquid/liquid (L/L) interface is the simplest and most promising model for understanding charge-transfer19 processes in biological systems.20,21 We have succeeded in embellishing a L/L interface with AAO to probe ion transport through ion channels in a confined nanospace in our previous studies.22,23 Even though H3PO4 can expand the size of the pore to speed up the transfer of ions and a protonated alumina surface can slow down the transfer of ions, the ion channel constructed formerly was a rigid nanochannel, which was quite different from biological ion channels. Therefore, we still need to construct nanochannel systems that have high similarity with biological ion channels. Xia's group has reported on the modification of AAO.24,25
This study was focused on the modification of a nanotube array with functional molecules that was assembled at a liquid–liquid interface to study ion transfer behavior by SECM (Scheme 1), which is a promising chemical model to investigate ion channels. It is worth mentioning that this chemical model provides a closer resemblance to biological ion channels because the molecular layer can serve as soft tissue. Furthermore, an acidic, neutral and basic pH of the solution resulted in complete, half and no protonation of the amino groups of the ion channel, respectively. The three different states, such as full, half and no protonation of the amino groups, showed a significant difference in the ion transport rates depending on the electrostatic interactions between the ions and surface groups, which acted simultaneously as pH-tunable asymmetric nanochannel systems and proton-sensitive gate to some extent. Based on this, we successfully constructed a controllable proton responsive nanochannel by functionalizing aluminium oxide nanotube array with 3-aminopropyltrimethoxysilane. The method introduced herein provides a new idea to investigate ion transport through channels.
 | ||
| Scheme 1 Representation of (a) ion transport through the ion channels, (b) the system used to investigate ion transport traversing the ion channels by SECM and (c) the ion channels modified with 3-aminopropyltrimethoxysilane. | ||
Such a strategy could provide a smart platform to explore the complex charge transfer reaction. A modified nanotube array can be used to assemble other molecules such as amino acids and proteins. Furthermore, it can also be applied in filtration, nanomedicine and biosensing.
Experimental
Materials
Sodium chloride (NaCl, AR), potassium chloride (KCl, AR), potassium ferrocyanide (K4Fe(CN)6, AR), potassium ferricyanide (K3Fe(CN)6, AR), and 1,2-dichloroethane (DCE, 99.8%, AR) were purchased from Beijing Chemical, China. Tetrabutylammonium tetraphenylborate (TBATPB, 99%, Aldrich), tetrabutylammonium chloride (TBACl, 99%, Aldrich), and tetramethylammonium chloride (TMACl, 99%, Aldrich) were used without purification. 3-Aminopropyltrimethoxysilane (APTMS) was obtained from Aladdin Reagent (China) Co. Ltd. Naphtho-15-crown-5 (N15C5) was synthesized via Pedersen's method and recrystallized three times from heptane.26 The AAO templates were made using a two-step anodic oxidation method27,28 and were left in water for about 12 h to ensure complete wetting before use. The organic phase (dichloroethane, DCE) was washed several times with deionized water before use. The aqueous solution was prepared using doubly distilled water obtained from a Milli-Q system (Milli-Q, Millipore Corp). Special cautions were taken for dealing with DCE and other hazardous chemicals.Apparatus and electrochemical measurements
A CHI-900 electrochemical workstation (CH Instruments Inc.) was connected to a personal computer, which was employed to record cyclic voltammograms and experimental approach curves. The nanopipets employed as the SECM tips were fabricated from borosilicate glass capillaries (o.d. = 2.0 mm, i.d. = 1.16 mm, L = 10 cm) using a laser based puller (made by our laboratory). Before each experiment, the nanopipets were checked using an olympus optical microscope to ensure that there was no trapped bubble. Aqueous solutions were infused into the pipets from the back with a small syringe (5 μL). As shown in Scheme 1, a three-electrode setup was employed with a nanopipet as the SECM tip, an Ag wire coated with AgTPBCl served as the reference electrode and a Pt wire served as the counter electrode. Ag/AgCl was inserted into the pipet as the reference electrode in the aqueous phase. The tip was filled with 0.3 M KCl aqueous solution from the back using a small syringe and biased at the potential wherein the facilitated K+ transfer reaction was diffusion-controlled. The droplet electrodes were used for the cyclic voltammograms. A drop of a certain volume of an aqueous solution containing KCl, K4Fe(CN)6 and K3Fe(CN)6 was transferred to the surface of a freshly polished platinum disk electrode (d = 2 mm) using a small syringe. The water-drop spread spontaneously across the surface of this platinum electrode and covered it completely. Furthermore, the electrode was turned over and immersed immediately into a DCE solution (of a certain volume) containing the ionophore (N15C5) and TBATPB salt. The experimental approach curves (iT–d curves) were obtained by moving the tip towards the bottom of the L/L interface and recording the tip current (iT) as a function of the tip interface separation distance (d). The coordinate of the L/L interface (d = 0) was decided from the sharp increase in iT that occurred when the tip touched the bottom DCE/W interface. All the experiments were carried out at room temperature (20 ± 2 °C).Preparation of AAO with different pore sizes
AAO templates with different pore sizes were fabricated via soaking AAO in 5 wt% phosphoric acid for different times.29 The pore-widening process could be easily carried out without changing the interpore distance, which was ideal for simulating the elasticity of ion channels found in living systems. As noted in our earlier report,23 the average size of the pores were measured and calculated, and were found to be 20, 30, 45 and 70 nm after pore widening treatment for a time of 10, 20, 30 and 40 min, respectively. Scanning electron microscopy (SEM) images of the AAO after different etching times in phosphoric acid are depicted in Fig. S1.† It could be observed that the average pore size was apparently different. Herein, the different pore-widening times were used to represent the average pore size due to the inhomogeneity of the pores.Surface modification
AAO templates with a pore diameter of about 30 nm were first cleaned as described elsewhere.30 The cleaned AAO was then immersed into a 10 mL acetone solution containing 10% 3-aminopropyltrimethoxysilane (APTMS) for about 12 h to graft the aminopropyl functional groups. Then, the excess APTMS was removed by washing the functionalized AAO with copious amounts of acetone. To guarantee the removal of adsorbed APTMS on the surface of the AAO, the washing process with acetone was repeated several times and then washed several times with deionized water. The templates were then dried under a stream of nitrogen to remove impurities and fluid. Fig. S2† shows the process used to graft the functional groups on the AAO.Characterization of the surface modification
The cleaned AAO before and after modified by APTMS was determined by SEM and energy dispersive spectroscopy (EDS). Fig. S3a and S4a† show the SEM images of the cross-section of the unmodified AAO and modified AAO, in which the nanotubes displayed uniform and vertically grown. Fig. S4b† shows the EDS characterization of the modified AAO. From the results it was observed that the APTMS has successfully modified the AAO nanotube array, as confirmed by the observable signal for nitrogen and silicon, whereas the EDS signal of nitrogen and silicon in the unmodified AAO was not detected in Fig. S3b.† The data shown in Tables 1 and 2 found in the ESI† plainly illustrated the EDS results of the AAO before and after modification in detail. These results show that ATPMS successfully modified the AAO nanotubes through the formation of Si–O bonds.Results and discussion
Mechanism of ion transport through the ion channels
The trans-membrane delivery of ions was caused by the concentration gradient between the intracellular and extracellular ions and the permeability of the biomembrane. There are three ways that a cell releases or takes up ions. As listed below: (i) passive transport: ions pass through the biomembrane facilitated by ionophores;31,32 (ii) active transport: ions pass through the biomembrane directly; and (iii) ion channels: ions pass through the biomembrane facilitated by specialized ion channels. In this study, the mechanism of ion transport belongs to the third way, which was attributed to the straight through-hole nanochannels of the AAO modified at the L/L interface.The transfer behavior of K+ and Na+ at the modified W/DCE interface was verified to involve two processes.22,23,32–34 The first process was the transfer by interfacial complexation (TIC) at the tip as shown below:

The second process was the transfer by interfacial dissociation (TID) at the bottom of W/DCE interface as shown below:

When the tip approaches the AAO nanotube array, M+ was released from the complex and transferred through the nanochannels into the aqueous solution, N15C5 was regenerated to its neutral form by interfacial dissociation and diffuses to the organic phase to produce the concentration gradient.
The following electrochemical cell (I) was employed for investigation of alkali metal ion transfer at the modified W/DCE interface facilitated by N15C5.
| Pt|1mMK4Fe(CN)6 + 1mMK3Fe(CN)6 + xMMCl(W)||5mMTBATPB + 1mMN15C5(DCE)|AgTPBCl|Ag Cell (1) |
When the concentration of the redox couple in the aqueous phase was in excess, the platinum–water interface was unpolarizable and acts as a reference/counter electrode for the aqueous phase. The current was then limited by the ion transfer rate through ion channels.
Ion transport traversing nanotubes array modified with surface functional groups
The protonation process of functional groups occurs in many biological processes. It was well documented that the pKa value of the amino groups in bulk solution is 10.6.35 However, the protonation of amino groups occurs in one step with a pK1/2 = 5.9 when groups were fixed on the inner surface of an alumina nanotube array using a silanization reaction.36 This difference could be ascribed to the confinement effect on the protonation of the amino groups in the nanochannel. They are not only affected by the neighboring amino groups but also by the opposite amino groups. In our previous report, the protonation of hydroxyl groups in porous anodic alumina was researched.23 The effect of pH was investigated by varying the pH from 6.5 to 8.0. Below a pH of 6.5, the alumina was protonated and has a positively charged surface (AlOH2+). Above a pH of 6.5, the alumina has a negatively charged surface (AlO−). The surface electrical properties significantly affect the diffusion behavior of charged species across these charged membranes. In the present study, AAO was decorated with APTMS containing amino groups, which might undergo a protonation process similar to that of hydroxyl groups. In addition, according to the titration curve for the protonation of the amino groups under the nanochannel confined conditions,36 the progress could be divided into three stages: (i) full-protonation state, (ii) half-protonation state, and (iii) non-protonation state. Below pH 5.5, the amino groups are protonated and have a positively charged surface (–NH3+). At about pH 5.5–7.0, the amino groups are partially protonated. Above pH 7.0, the amino groups are not charged and exist in their non-protonated form (–NH2). The ion channel surface was positively charged and attracts anions, whereas cations are repelled. Therefore, an ion confined-diffusion region was formed near a charged surface in contact with an electrolyte. The surface electrical properties could modulate the transportation of charged electroactive probes through the nanochannels.37,38 Scheme 2 shows the distinct diffusion behavior of ions across the ion channels in the three different states of the surface amino groups. The diffusion of ions through the charged ion channels could be divided into two different contributions: (i) the confined diffusion region and (ii) the free diffusion region. In this assay, the pH values of the aqueous solutions selected herein were 5.0, 6.0, and 8.0, respectively, which represented the three different protonation states. | ||
| Scheme 2 A schematic of the diffusion of target ions across a functionalized alumina nanotube array. | ||
Herein, the experimental SECM approach curves of the L/L-modified interface used the Bard's formula.39 The steady-state current obtained was consistent with the asymmetric diffusion regime, which was formed by the specific shape of the pipet and the mechanism of the facilitated ion transfer (IT).40,41 The driving force was provided by the voltage, which was applied between the pipet and the reference electrode for the IT process. With the concentration of K+ or Na+ in the aqueous phase inside a pipet higher than the concentration of N15C5 in the organic phase, the current was seriously limited by the diffusion of N15C5 to the pipet orifice. Assuming that the pipet orifice was disk-shaped, calculation of the steady-state diffusion limiting current used the equation as follows:42–45
| id = 4nfaDc | (1) |
The cyclic voltammograms and approach curves for K+ and Na+ transfer through the ion channels at the W/DCE interface at different pH are shown in Fig. 1. Moreover, different peak currents (i) were achieved under different pH conditions as shown in Fig. 1a, pH = 5, i = 0.5466 × 10−7 A; pH = 6, i = 1.0713 × 10−7 A; pH = 8, i = 1.4758 × 10−7 A. From Fig. 1c, pH = 5, i = 0.5808 × 10−7 A; pH = 6, i = 0.7851 × 10−7 A; pH = 8, i = 0.9989 × 10−7 A. It was clearly observed that the peak current decreased in the steady-state current upon changing the degree of protonation of the amino groups from the non-protonation to full-protonation state. The highest current response was observed in non-protonation state of amino groups. Good agreement between the obtained results and theoretical hypothesis confirmed the enhanced confinement of motion in the confined-diffusion region was attributed to the increased surface charge density observed at lower pH values. Fig. 2 displays the transport rate constants obtained from experimental approach curves. As expected, in the non-protonation state, the ion transport rate increased for potassium ions and sodium ions (7.3 times compared with the full-protonation state). The ion transport rate of potassium ions in the non-protonation state increased to 0.0081 cm s−1, which was much higher than that in the full-protonation state. Therefore, the ion transfer in the full-protonation state of amino groups could be reasonably neglected. There exists a certain degree of similarity between gated channels in vivo and the proposed nanochannel. To date, it is difficult to achieve the goal that no ions transport through the channel; it is still a challenge to control precisely the density of the grafted functional molecules in terms of surface coverage.
 | ||
| Fig. 1 The influence of pH of the aqueous solution on alkali metal ion transfer. Cyclic voltammograms for (a) K+ and (c) Na+ transfer facilitated by N15C5 through ion channels at different pH values in the range from 5.0 to 8.0. Experimental approach curves (dot) fitted with the theoretical values (line) for (b) K+ and (d) Na+ transfer. The concentration of K+ and Na+ was 0.3 M and the concentration of N15C5 was 1 mM. The sweep rate was 50 mV s−1. | ||
 | ||
| Fig. 2 Transfer rate constant vs. pH. | ||
The transfer rate of ions through the nanochannels varies with the thickness of the confined diffusion region. The thickness of confined transport region greatly depends on the Debye length rD, which has the formula:
 | (2) |
 | ||
| Fig. 3 Cyclic voltammograms for (a) K+ and (c) Na+ transfer facilitated by N15C5 through ion channels. Experimental approach curves (dots) fitted with the theoretical values (lines) of (b) K+ and (d) Na+ transfer. The ionic strength of K+ varies from 0.1 to 0.5 M and the concentration of N15C5 was 1 mM. The sweep rate was 50 mV s−1. | ||
The voltammetric behavior obeys the revised Randles–Sevcik equation shown below as follows:23
| ip = 0.4463 × 10−3 × n3/2F3/2(RT)−1/2mπ(rp − rD)2DM+1/2cM+v1/2 | (3) |
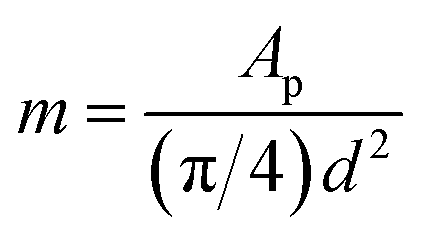 | (4) |
The kinetic parameters of the transfer reactions could be evaluated using the three-point method, which can be found from the literature to obtain values of ΔE1/4 and ΔE3/4.50 The standard rate constant (k0) is given by the equation as follows:
 | (5) |
The effect of the size of the nanochannels
From what has been discussed above, it could be said that the confined-diffusion region plays an important role in regulating ion transport through ion channels. The contribution of this region to the total ion transfer strongly depends on the pore size and therefore further study was undertaken to discuss the effect of the pore size. A family of experimental SECM approach curves which fitted with the theoretical values at different etching times (t = 0 min, 10 min, 20 min, 30 min, and 40 min) in the neutral pH environment similar to a natural organism are shown in Fig. 4. The transfer rate constants increased from 0.0044 to 0.0180 cm s−1 with increasing pore diameters in the range of 20–70 nm (Fig. 5). The CVs at the four different etching times (t = 10 min, 20 min, 30 min and 40 min) showed that the current signal obtained across ion channels with large pores was stronger than that with small pores. They were consistent with the results obtained from the SECM approach curves. The results shown above could be ascribed to the thickness of the confined-diffusion region and its percentage in the pore cross-section.23 When pore size was very small, the percentage of the confined-diffusion region to the pore cross-section was increased. Thus, the pathway of free-diffusion of K+ or Na+ was decreased, which resulted in a decreased mass transportation of ions through the nanochannels. On the contrary, mass transportation of K+ or Na+ through nanochannels was expected to increase with increasing pore diameter, which caused a stronger current response, i.e., it was easier for K+ to pass through the ion channels with big pores. Therefore, these observations indicate that pore size of the nanochannel plays an important role in the regulation of ion transport.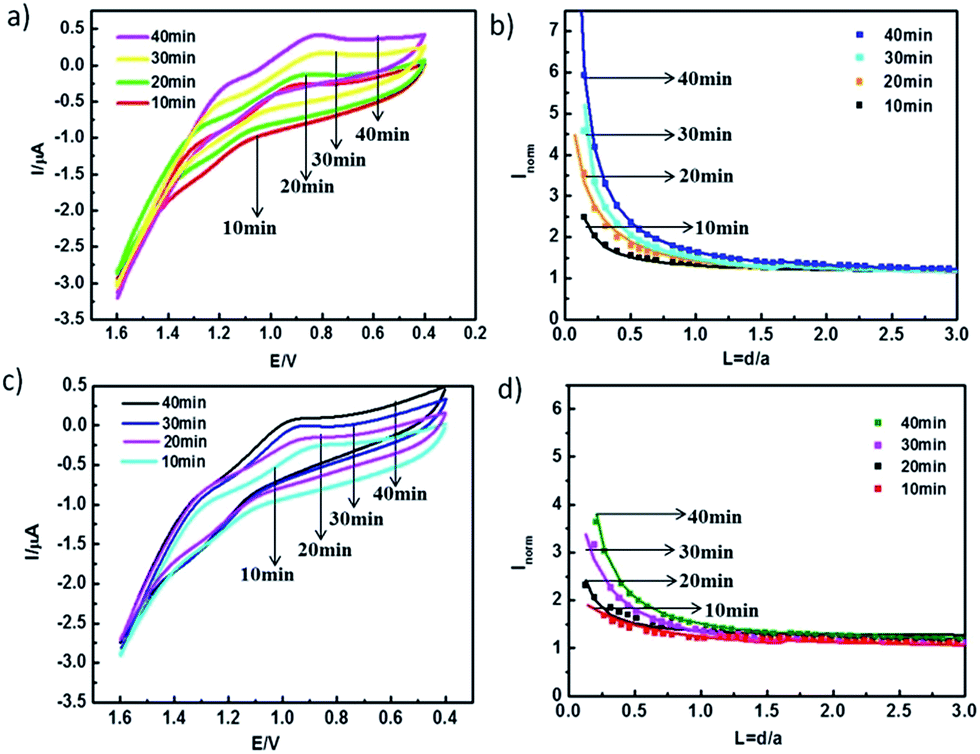 | ||
| Fig. 4 The influence of the pore diameter of AAO on ion transfer. Cyclic voltammograms for (a) K+ and (c) Na+ transfer. Experimental approach curves (dots) fitted with the theoretical values (lines) of (b) K+ and (d) Na+ transfer facilitated by N15C5 through ion channels with different pore diameters. The time of pore-widening in 5 wt% H3PO4 was 10 min, 20 min, 30 min, and 40 min, respectively, and the concentration of N15C5 was 1 mM. The sweep rate was 50 mV s−1. | ||
 | ||
| Fig. 5 The transfer rate constant vs. pore widening time (pore diameter of AAO). | ||
From the whole course of the experiment, the pH, ion concentration and pore size were found to impact the transfer behavior of the ions. When the ion concentration and pore size remained unchanged, the different protonation states of the amino group, which were a result of the variation in the pH of the solution in the confined nanochannel could distinctly regulate ion transport. Moreover, the ion concentration and pore size affect the thickness of the confined diffusion region. When the pH remained unchanged, the peak current increased with increasing ion concentration and pore size. Those conditions are of great significance to researching ion transfer in living organisms.
Conclusions
In summary, a new model used to investigate the ions transfer across controllable ion channels was constructed by decorating the interior surface of a nanotube array with functional molecules. Molecular layers assembled on the inner wall of the nanotube array can play the part of organic soft tissue and was more similar to ion channels found in biological systems. The different protonation states of the amino groups in the confined nanochannel can distinctly regulate ion transport and was very similar to the switching effect of ion channels in vivo. To date, it was difficult to achieve the goal of switching the channel completely off because there exists the electrostatic repulsion between the modified amino groups, but most important is the non-uniformity of the adjacent pore size. Therefore, we aim to overcome the abovementioned shortcomings in our future studies. However, the nanotube array constructed herein can better imply the ion transfer kinetics and the further study and development of these ion channels will allow their use in more complex biomimetic materials in various nanotechnology and biomechanical applications.Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grant No. 21327005, 21575115); the Program for Chang Jiang Scholars and Innovative Research Team, Ministry of Education, China. (Grant No. IRT1283); the Program for Innovative Research Group of Gansu Province, China (Grant No. 1210RJIA001) and the Program of Innovation and Entrepreneurial for Talent, Lan Zhou, Gansu Province, China (Grant No. 2014-RC-39).Notes and references
- T. Sumikama, S. Saito and I. Ohmine, J. Phys. Chem. B, 2006, 110, 20671–20677 CrossRef CAS PubMed.
- A. Felipe, R. Vicente, N. Villalonga, M. Roura-Ferrer, R. Martínez-Mármol, L. Solé, J. C. Ferreres and E. Condom, Cancer Detect. Prev., 2006, 30, 375–385 CrossRef CAS PubMed.
- F. I. Valiyaveetil, Y. Zhou and R. MacKinnon, Biochemistry, 2002, 41, 10771–10777 CrossRef CAS PubMed.
- A. D. Wickenden, Pharmacol. Ther., 2002, 94, 157–182 CrossRef CAS PubMed.
- F. Lehmann-horn and K. Jurkat-rott, Physiol. Rev., 1999, 79, 1317–1372 CAS.
- J. Patlak, Physiol. Rev., 1991, 71, 1047–1080 CAS.
- B. Martinac, J. Cell Sci., 2004, 117, 2449–2460 CrossRef CAS PubMed.
- A. W. Catterall, Trends Neurosci., 1993, 16, 500–506 CrossRef PubMed.
- C. Rue and P. B. Armentrout, J. Phys. Chem. A, 2002, 106, 9788–9797 CrossRef CAS.
- I. H. Shrivastava and M. S. Sansom, Biophys. J., 2000, 78, 557–570 CrossRef CAS PubMed.
- A. Cha, G. E. Snyder, P. R. Selvin and F. Bezanilla, Nature, 1999, 402, 809–813 CrossRef CAS PubMed.
- W. A. Schmalhofer, M. Sanchez, G. Dai, A. Dewan, L. Secades, M. Hanner, H. G. Knaus, O. B. McManus, M. Kohler and G. J. Kaczorowski, Biochemistry, 2005, 44, 10135–10144 CrossRef CAS PubMed.
- X. Hou, W. Guo and L. Jiang, Chem. Soc. Rev., 2011, 40, 2385–2401 RSC.
- F. Dehez, M. Tarek and C. Chipot, J. Phys. Chem. B, 2007, 111, 10633–10635 CrossRef CAS PubMed.
- C. D. Assouma, A. Crochet, Y. Chérémond, B. K. Giese and M. Fromm, Angew. Chem., Int. Ed., 2013, 52, 4682–4685 CrossRef CAS PubMed.
- P. Apel, Radiat. Meas., 2001, 34, 559–566 CrossRef CAS.
- Z. X. Lu, A. Namboodiri and M. M. Collinson, ACS Nano, 2008, 2, 993–999 CrossRef CAS PubMed.
- W. Chen, J. S. Wu and X. H. Xia, ACS Nano, 2008, 2, 959–965 CrossRef CAS PubMed.
- T. J. Stockmann and Z. F. Ding, Phys. Chem. Chem. Phys., 2012, 14, 13949–13954 RSC.
- T. Ohkouchi, T. Kakutani and M. Senda, Bioelectrochem. Bioenerg., 1991, 25, 71–80 CrossRef CAS.
- Z. F. Ding, B. M. Quinn and A. J. Bard, J. Phys. Chem. B, 2001, 105, 6367–6374 CrossRef CAS.
- X. Q. Lu, T. X. Wang, X. B. Zhou, Y. Li, B. W. Wu and X. H. Liu, J. Phys. Chem. C, 2011, 115, 4800–4805 CAS.
- H. Xia, D. D. Qin, X. B. Zhou, X. H. Liu and X. Q. Lu, J. Phys. Chem. C, 2013, 117, 23522–23528 CAS.
- H. L. Gao, C. Y. Li, F. X. Ma, K. Wang, J. J. Xu, H. Y. Chen and X. H. Xia, Phys. Chem. Chem. Phys., 2012, 14, 9460–9467 RSC.
- S. J. Li, J. Li, K. Wang, C. Wang, J. J. Xu, H. Y. Chen, X. H. Xia and Q. Huo, ACS Nano, 2010, 11, 6417–6424 CrossRef PubMed.
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 7017–7036 CrossRef CAS.
- D. D. Li, L. Zhao, C. H. Jiang and J. G. Lu, Nano Lett., 2010, 10, 2766–2771 CrossRef CAS PubMed.
- J. H. Yuan, F. Y. He, D. C. Sun and X. H. Xia, Chem. Mater., 2004, 16, 1841–1844 CrossRef CAS.
- J. Zhang, J. E. Kielbasa and D. L. Carroll, Mater. Chem. Phys., 2010, 122, 295–300 CrossRef CAS.
- H. L. Gao, H. Zhang, C. Y. Li and X. H. Xia, Electrochim. Acta, 2013, 110, 159–163 CrossRef CAS.
- D. P. Zhan, S. N. Mao, Q. Zhao, Z. Chen, H. Hu, P. Jing, M. Q. Zhang, Z. W. Zhu and Y. H. Shao, Anal. Chem., 2004, 76, 4128–4136 CrossRef CAS PubMed.
- H. H. Girault, J. O. M. Bockris, B. E. Conway and R. E. White, Modern Aspects of Electrochemistry, Plenum, New York, 1993, vol. 25, p. 1 Search PubMed.
- Y. H. Shao, M. D. Osborne and H. H. Girault, J. Electroanal. Chem., 1991, 318, 101–109 CrossRef CAS.
- J. Zhang and P. R. Unwin, Langmuir, 2002, 18, 2313–2318 CrossRef CAS.
- H. Zhang, H. X. He, J. Wang, T. Mu and Z. F. Liu, Appl. Phys. A, 1998, 66, S269–S271 CrossRef CAS.
- H. L. Gao, K. Zhou, C. Wang, S. Li, H. Zhang and X. H. Xia, J. Electrochem., 2012, 18, 229–234 CAS.
- E. A. Bluhm, E. Bauer, R. M. Chamberlin, K. D. Abney, J. S. Young and G. D. Jarvinen, Langmuir, 1999, 15, 8668–8672 CrossRef CAS.
- W. Chen, Z. Q. Wu, X. H. Xia, J. J. Xu and H. Y. Chen, Angew. Chem., Int. Ed., 2010, 49, 7943–7947 CrossRef CAS PubMed.
- C. Wei, A. J. Bard and M. V. Mirkin, J. Phys. Chem., 1995, 99, 16033–16042 CrossRef CAS.
- Y. H. Shao, M. D. Osborne and H. H. Girault, J. Electroanal. Chem., 1991, 318, 101–109 CrossRef CAS.
- Y. H. Shao and M. V. Mirkin, Anal. Chem., 1997, 69, 1627–1634 CrossRef CAS.
- P. D. Beattie, A. Delay and H. H. Girault, J. Electroanal. Chem., 1995, 380, 167–175 CrossRef.
- M. Tsionsky, J. Zhou, S. Amemiya, F. R. F. Fan, A. J. Bard and R. A. W. Dryfe, Anal. Chem., 1999, 71, 4300–4305 CrossRef CAS PubMed.
- T. Solomont and A. J. Bard, J. Phys. Chem., 1995, 99, 17487–17489 CrossRef.
- B. Su, S. Zhang, Y. H. Yuan, J. Guo, L. Gan and Y. Shao, Anal. Chem., 2001, 74, 373–378 CrossRef.
- Y. H. Shao and M. V. Mirkin, Anal. Chem., 1998, 70, 3155–3161 CrossRef CAS PubMed.
- F. Li, Y. Chen, P. Sun, M. Zhang, Z. Gao, D. P. Zhan and Y. H. Shao, J. Phys. Chem. B, 2004, 108, 3295–3302 CrossRef CAS.
- A. Stewart, G. Taylor, H. H. Girault and J. McAleer, J. Electroanal. Chem., 1990, 296, 491–515 CrossRef CAS.
- A. Belwalkar, E. Grasing, W. V. Geertruyden, Z. Huang and W. Misiolek, J. Membr. Sci., 2008, 319, 192–198 CrossRef CAS PubMed.
- M. V. Mirkin and A. J. Bard, Anal. Chem., 1992, 64, 2293–2302 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: SEM images of AAO, functionalization of the inner walls of AAO, SEM and EDS image of cross-section of AAO nanotube array modified with APTMS. See DOI: 10.1039/c5ra25114a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |