
IDO as a drug target for cancer immunotherapy: recent developments in IDO inhibitors discovery
Abstract
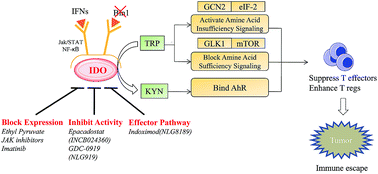
The indoleamine 2,3-dioxygenase (IDO) mediated kynurenine pathway of tryptophan degradation is identified as an important immune effector pathway in tumor cells to escape a potentially effective immune response. IDO affects the differentiation and proliferation of T cells, triggering downstream signaling through GCN2, mTOR and AhR. Therefore, IDO is an attractive target for cancer immunotherapy. IDO inhibitors exhibit potent anticancer activities and might be very useful in combination with chemotherapy, radiotherapy or immunotherapy to trigger the rapid regression of aggressive tumors. However, the development of IDO pharmacological inhibitors has been a challenging work. This review highlights recent advances (2010–2015) in research related to the role of IDO in immune escape and pathogenic inflammation in cancer, and novel small-molecule IDO inhibitors with an emphasis on their chemical structures and modes of action.
 Please wait while we load your content...
Please wait while we load your content...

