
Enhanced purification of carbon nanotubes by microwave and chlorine cleaning procedures
Abstract
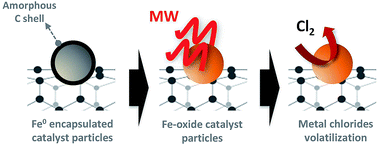
A new two-step purification method of carbon nanotubes (CNTs) involving a microwave treatment followed by a gas-phase chlorination process is reported. The significant advantage of this method over conventional cleaning carbon nanotubes procedures is that under microwave treatment in air, the carbon shells that encase the residual metal catalyst particles are removed and the metallic iron is exposed and subsequently oxidized making it accessible for chemical removal. The products from microwave and chlorine treatment have been characterized by TG/DTA, SEM, TEM, EDX, XPS, and Raman spectroscopy. The oxidation state of the iron residue is observed to change from Fe(0) to Fe(II)/Fe(III) after microwave treatment and atmospheric exposure. The effects of the duration and number of microwave exposures has been investigated. This rapid and effective microwave step favours the subsequent chlorination treatment enabling a more effective cleaning procedure to take place, yielding higher purity single- and multi-walled CNTs.
 Please wait while we load your content...
Please wait while we load your content...

