
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Vanadium(V) phenolate complexes for ring opening homo- and co-polymerisation of ε-caprolactone, L-lactide and rac-lactide†
Feijie
Ge
a,
Yi
Dan
a,
Yahya
Al-Khafaji
b,
Timothy J.
Prior
b,
Long
Jiang
*a,
Mark R. J.
Elsegood
c and
Carl
Redshaw
*b
aState Key Laboratory of Polymer Materials Engineering of China (Sichuan University), Polymer Research Institute of Sichuan University, Chengdu 610065, Sichuan, China. E-mail: jianglong@scu.edu.cn; Fax: +86-28-85402465; Tel: +86-28-85407286
bDepartment of Chemistry, University of Hull, Hull, HU6 7RX, UK. E-mail: C.Redshaw@Hull.ac.uk; Fax: +44 (0)1482 466410; Tel: +44 (0)1482 465219
cChemistry Department, Loughborough University, Leicestershire LE11 3TU, UK
First published on 22nd December 2015
Abstract
The vanadyl complexes [VO(OtBu)L1] (1) and {[VO(OiPr)]2(μ-p-L2p)} (2) {[VO(OR)]2(μ-p-L2m)} (R = iPr 3, tBu 4) have been prepared from [VO(OR)3] (R = nPr, iPr or tBu) and the respective phenol, namely 2,2′-ethylidenebis(4,6-di-tert-butylphenol) (L1H2) or α,α,α′,α′-tetra(3,5-di-tert-butyl-2-hydroxyphenyl–p/m-)xylene-para-tetraphenol (L2p/mH4). For comparative studies, the known complexes [VO(μ-OnPr)L1]2 (I), [VOL3]2 (II) (L3H3 = 2,6-bis(3,5-di-tert-butyl-2-hydroxybenzyl)-4-tert-butylphenol) were prepared. An imido complex {[VCl(Np-tolyl)(NCMe)]2(μ-p-L2p)} (5) has been prepared following work-up from [V(Np-tolyl)Cl3], L2pH4 and Et3N. The molecular structures of complexes 1–5 are reported. Complexes 1–5 and I and II have been screened for their ability to ring open polymerise ε-caprolactone, L-lactide or rac-lactide with and without solvent present. The co-polymerization of ε-caprolactone with L-lactide or rac-lactide afforded co-polymers with low lactide content; the reverse addition was ineffective.
Introduction
As a biodegradable polyester that has the potential for replacing traditional polymers, polylactide (PLA) has attracted much attention in recent years.1 ε-Caprolactone too is attracting interest as a precursor to biodegradable polycaprolactone (PCL) polymers.2 Both types of polymer are readily formed via ring opening polymerization (ROP) of the respective monomer catalyzed by metal complexes, which can be a living process thereby allowing for good control. Given the bio-applications of PLA and PCL type polymers, there is a drive to develop syntheses using new catalysts that offer advantages over the established stannous octoate,3 yet which possess low toxicity. Furthermore, the properties associated with PLA and PCL can be quite different, as typified by their differences in elasticity, and so there is a drive to produce ε-Cl/LA co-polymers of varying composition (and properties).One metal attracting attention in polymerisation catalysis is vanadium.4 Given the toxicity associated with this metal is relatively low,5 we have embarked upon a program to screen various vanadium systems for their ability to deliver, via ROP, biodegradable polymers with desirable properties. We note that reports on the use of group 5 complexes for the ROP of cyclic esters are scant.6 Herein, we investigate the potential of vanadyl complexes with ligands derived from di-(L1H2) or tetra-phenols (L2p/mH4) and for comparative studies the tri-phenol (L3H3) (see Scheme 1) for the ROP of both L-lactide and ε-caprolactone and the co-polymerisation thereof, and report the effects of the structures of the complexes on the properties of the final polymeric products. Such chelating phenoxide ligation has proved useful in olefin polymerisation,4 but their use in the ROP of lactides and lactones, particularly that of the tri- and tetra-phenolic ligand sets, is rather limited.6d,7,8 Use of a tetra-phenolate ligand set, both meta and para forms, also allowed us to probe for possible cooperative effects, viz.1versus2–4. With this in mind, the crystal structures of the monomeric vanadyl complex [VO(OtBu)L1] (1) and the dinuclear vanadyl complexes {[VO(OiPr)]2(μ-p-L2p)} (2), {[VO(OR)]2(μ-p-L2m)} (R = iPr 3, tBu 4) are also presented; the molecular structures of I and II have been reported elsewhere.9
 | ||
| Scheme 1 Vanadyl complexes screened herein (Ar = p-tolyl, L = MeCN). | ||
Results and discussion
Vanadyl phenolate complexes
Interaction of [VO(OtBu)3] with the ethylidene-bridged di-phenol 2,2′-ethylidenebis(4,6-di-tert-butylphenol), 2,2′-CH3CH[4,6-(tBu)2C6H2OH]2 (LH2) in refluxing toluene afforded, after workup, the monomeric complex [VO(OtBu)L] (1) in good isolated yield (ca. 76%). Complex 1 is presumed to form via displacement of two molecules of tert-butanol in a similar fashion reported for related n-propoxide complexes.9 In the IR of 1, there is a strong stretch at 1003 cm−1 assigned to the ν(V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) group. Crystals of 1 suitable for X-ray diffraction were readily grown from a saturated acetonitrile solution at 0 °C. The structure of 1 is shown in Fig. 1 (for ORTEP, see Fig. S1†), with selected bond lengths and angles given in the caption; crystal structure data are given in Table S2.† The vanadium centre adopts a distorted tetrahedral environment with angles varying from ideal in the range 107.7(2) to 112.1(2)°. The chelating ligand forms an eight membered metallocycle adopting a flattened chair conformation, with a bite angle of 110.1(2)°, which is somewhat larger than that found in the monomeric vanadyl complex {VOCl[2,2′-CH2(4-Me,6-tBuC6H2O)2]} [106.9(2)°] and the dimeric complex [VO(OnPr)L1]2 [94.49(10)°].9a,10 The V–O bond lengths to the bisphenolate ligand [1.789(5) and 1.783(5) Å] are typical of those previously observed for vanadium aryloxides,9,10 whilst the V–O alkoxide distance [1.739(5) Å] is shorter than those typically observed in alkoxy vanadium complexes, but similar to that reported in the monomeric imido vanadium complex [V(NAr)(OtBu)L] [1.738(2) Å] (Ar = p-ClC6H4).11 The alkoxide ligand is best described as bent with a V(1)–O(4)–C(40) angle of 145.9(5)°, which is slightly smaller than the analogous angle in the imido complexes [V(NAr)(OtBu)L] [146.77(12)–151.9(2)°] (Ar = p-ClC6H4, p-tolyl).11
O) group. Crystals of 1 suitable for X-ray diffraction were readily grown from a saturated acetonitrile solution at 0 °C. The structure of 1 is shown in Fig. 1 (for ORTEP, see Fig. S1†), with selected bond lengths and angles given in the caption; crystal structure data are given in Table S2.† The vanadium centre adopts a distorted tetrahedral environment with angles varying from ideal in the range 107.7(2) to 112.1(2)°. The chelating ligand forms an eight membered metallocycle adopting a flattened chair conformation, with a bite angle of 110.1(2)°, which is somewhat larger than that found in the monomeric vanadyl complex {VOCl[2,2′-CH2(4-Me,6-tBuC6H2O)2]} [106.9(2)°] and the dimeric complex [VO(OnPr)L1]2 [94.49(10)°].9a,10 The V–O bond lengths to the bisphenolate ligand [1.789(5) and 1.783(5) Å] are typical of those previously observed for vanadium aryloxides,9,10 whilst the V–O alkoxide distance [1.739(5) Å] is shorter than those typically observed in alkoxy vanadium complexes, but similar to that reported in the monomeric imido vanadium complex [V(NAr)(OtBu)L] [1.738(2) Å] (Ar = p-ClC6H4).11 The alkoxide ligand is best described as bent with a V(1)–O(4)–C(40) angle of 145.9(5)°, which is slightly smaller than the analogous angle in the imido complexes [V(NAr)(OtBu)L] [146.77(12)–151.9(2)°] (Ar = p-ClC6H4, p-tolyl).11
 | ||
| Fig. 1 Molecular structure of 1. Selected bond lengths (Å) and angles (°): V(1)–O(1) 1.789(5), V(1)–O(2) 1.783(5), V(1)–O(3) 1.581(5),V(1)–O(4) 1.739(5); O(1)–V(1)–O(2) 110.1(2), V(1)–O(1)–C(1) 124.4(4), V(1)–O(2)–C(17) 130.9(4), V(1)–O(4)–C(41) 145.9(5). | ||
| Bond lengths (Å)/angles (°) | 3 | 4·2CH2Cl2 | 4·3CH2Cl2 |
|---|---|---|---|
| V1–O(phenolate) | 1.790(2) | 1.7908(15) | 1.790(6) |
| 1.790(2) | 1.7963(15) | 1.806(5) | |
| V1O(vanadyl) |
1.581(3) | 1.5890(16) | 1.577(6) |
| V1–O(alkoxide) | 1.733(3) | 1.7298(16) | 1.736(6) |
| V2–O(phenolate) | 1.791(2) | 1.7891(16) | 1.792(6) |
| 1.793(2) | 1.7904(15) | 1.796(5) | |
| V2O(vanadyl) |
1.584(3) | 1.5908(17) | 1.572(7) |
| V2–O(alkoxide) | 1.734(3) | 1.7323(16) | 1.735(6) |
| O1–V1–O2 | 111.31(7) | ||
| O2–V1–O5 | 106.92(8) | ||
| O5–V1–O6 | 112.95(8) | ||
| V1–O1–C1 | 126.91(13) | ||
| V1–O16–C1 | 126.91(13) | ||
| V1–O6–C65 | 143.63(15) |
Extension of this synthetic methodology to the tetra-phenol α,α,α′,α′-tetra(3,5-di-tert-butyl-2-hydroxyphenyl–p-)xylene-para-tetra-phenol (L2pH4) afforded the dinuclear complex {[VO(OiPr)]2(μ-p-L2p)} (2) in good yield.
Crystals of 2 suitable for X-ray diffraction were readily grown from a saturated dichloromethane solution at 0 °C. The structure of 2 is shown in Fig. 2 (for ORTEP, see Fig. S2†), with selected bond lengths and angles given in the caption; crystal structure data are given in Table 5. The tetra-phenolate ligand is centrosymmetric with one vanadyl cation bound above the plane of the central aromatic ring and one beneath. The separation of these two identical metal centres is 11.756 Å. Each vanadium centre can be described as adopting a pseudo tetrahedral geometry. The bite angle formed by the tetra-phenolate at each vanadium is 109.4(2)°, which is slightly smaller than that observed for 1 (110.1(2)°) and for the recently reported alkoxide complexes {[VO(OR)]2(μ-p-L2p)} [R = nPr, 111.73(7)°; tBu, 112.0(2)°]; and the metallocycle adopts a flattened boat conformation. The isopropoxide ligand can be described as bent with a V2–O8–C68 angle of 130.1(5)°.
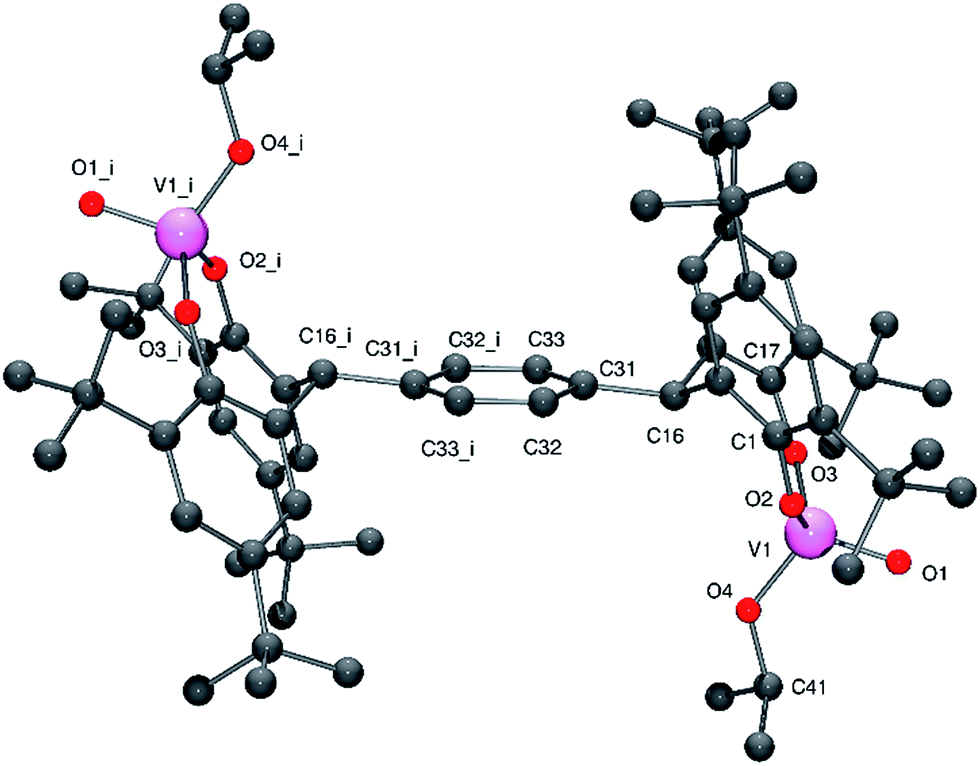 | ||
| Fig. 2 Molecular structure of 2. Selected bond lengths (Å) and angles (°): V1–O1 1.575(4), V1–O2 1.777(4), V1–O3 1.750(5),V1–O4 1.759(5); O2–V1–O3 109.4(2), V1–O2–C1 139.7(5), V1–O3–C17 151.7(4), V1–O4–C40 130.1(5). | ||
Similar use of the meta tetra-phenol α,α,α′,α′-tetra(3,5-di-tert-butyl-2-hydroxyphenyl–m-)xylene-meta-tetra-phenol (L2mH4) with [VO(OR)3] (R = nPr or tBu) afforded the dinuclear complexes {[VO(OR)]2(μ-m-L2m)} (R = iPr 3, tBu 4) in good yield. Crystals of 3 and of 4 suitable for an X-ray diffraction studies were obtained on cooling (to −20 °C) their respective saturated dichloromethane solutions. The molecular structure of 3·2CH2Cl2 is shown in Fig. 3 (for ORTEP, see Fig. S3†), with selected bond lengths (Å) and angles (°) given in Table 1 where they are compared with those of 4·2CH2Cl2 and 4·3CH2Cl2.
 | ||
| Fig. 3 Molecular structure of complex 3·2CH2Cl2, indicating the atom numbering scheme. Hydrogen atoms and solvent molecules of crystallisation have been removed for clarity. | ||
In 3, the vanadium centres can be described as adopting a pseudo tetrahedral geometry. The bite angle formed by the tetra-phenolate at each vanadium is 110.55(11)°, which is slightly larger than that observed for 1; again the metallocycle adopts a chair-boat conformation. The iso-propoxide ligand can be described as bent with a V2–O8–C68 angle of 140.5(3)°; the V–O iso-propoxide bond lengths are similar to those observed elsewhere.12
For 4·2CH2Cl2, there is one molecule of the complex and two molecules of CH2Cl2 (modelled by the Platon SQUEEZE procedure) in the asymmetric unit.13 Each vanadium center adopts a pseudo-tetrahedral geometry (see Fig. 4; for ORTEP, see Fig. S4†), with bond angles in the range 106.92(8)–112.95(8)°; the bite angle of the chelate is 111.31(7)°. The tert-butoxide ligand is somewhat bent [V1–O6–C65 = 143.63(15)°], with a slightly larger angle than that observed in the iso-propoxide 3 and presumably reflects the greater steric bulk of the tert-butoxide. The molecules pack in layers, however there is no significant interaction between the layers.
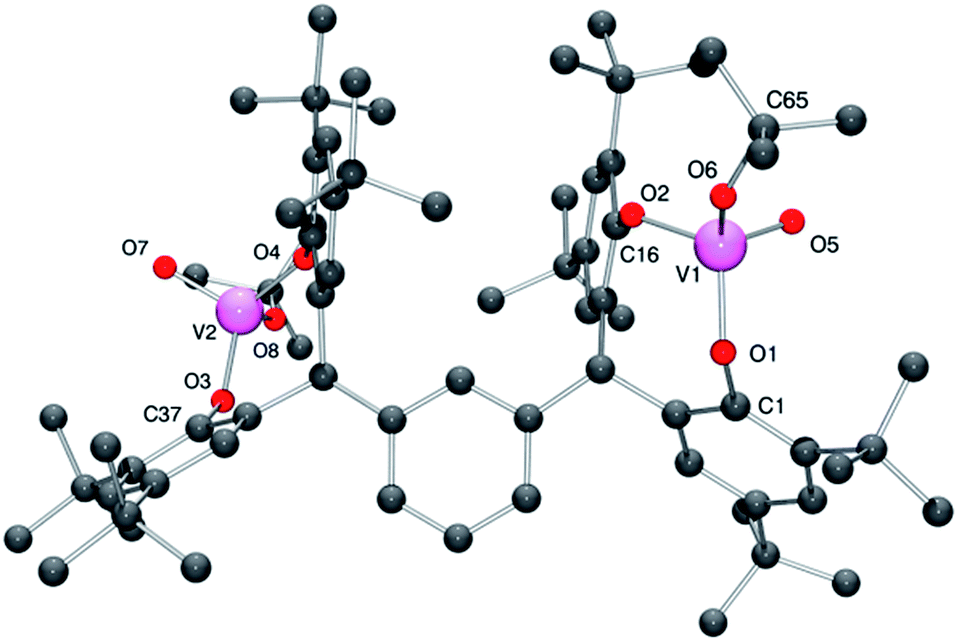 | ||
| Fig. 4 Molecular structure of complex 4·2CH2Cl2, indicating the atom numbering scheme. Hydrogen atoms and solvent molecules of crystallisation have been removed for clarity. | ||
From a repeated synthesis of 4, a different solvate was obtained, namely 4·3CH2Cl2. In the molecular structure of 4·3CH2Cl2, determined using synchrotron radiation,14 there is one well-defined dichloromethane which is involved in intramolecular interactions (see Fig. 5, for ORTEP, see Fig. S5†). In particular, there is a C–H⋯O H-bond to the oxo group O7 (see Table S1 in ESI† for geometry) and a C–H⋯π interaction with the aromatic ring C51 > C56 {H(73B)⋯ring centroid = 2.56(2) Å}.
 | ||
| Fig. 5 Molecular structure of complex 4·3CH2Cl2, indicating the atom numbering scheme. Hydrogen atoms except those on the CH2Cl2 have been removed for clarity. | ||
The known vanadyl complexes [VO(μ-OnPr)L1]2 (I), [VOL3]2 (II) were prepared as described previously (L3H3 = 2,6-bis(3,5-di-tert-butyl-2-hydroxybenzyl)-4-tert-butylphenol) via the reaction of [VO(OnPr)3] and the respective chelating phenol.9
Vanadium imido phenolate complex
Given the oxo group is isoelectronic with the imido group, we extended the studies to the reaction of [V(Np-MeC6H4)Cl3],15 with p-L2pH4 in the presence of Et3N. Following work-up (extraction into MeCN), the red/brown imido complex {[V(Np-MeC6H4)(NCMe)Cl]2(μ-p-L2p)}·2MeCN (5·2MeCN) was isolated in good yield. Single crystals of 5, obtained on prolonged standing at ambient temperature, were subjected to an X-ray diffraction study. The structure of 5 is shown in Fig. 6 (for ORTEP, see Fig. S6†), with selected bond lengths and angles given in the caption. The geometry at each vanadium is best described as trigonal bipyramidal with the imido and MeCN groups occupying axial positions [N1–V1–N2 178.74(14)°]. Bond angles are in the range 111.59(11)–122.47(9)°, with the largest equatorial deviation associated with the angle subtended at the metal by the phenolic oxygen centres. The imido ligand has the geometrical parameters associated with a linear imido function [V1–N1 1.670(3) Å; V1–N1–C40 170.5(3)°]. The structure of 5 closely resembles that of the recently reported complexes {[V(NAr)(THF)Cl]2(μ-p-L2p)} (Ar = p-MeC6H4, p-CF3C6H4), in which THF occupies one of the axial position at the metal as opposed to MeCN in 5.4d | ||
| Fig. 6 Molecular structure of complex 5·2MeCN, indicating the atom numbering scheme. Hydrogen atoms and solvent molecules of crystallisation have been removed for clarity. Selected bond lengths (Å) and angles (°): V1–N1 1.670(3), V1–O1 1.818(3), V1–O2 1.831(2), V1–Cl1 2.2609(11); O1–V1–N1 98.33(13), O1–V1–O2 111.59(11), Cl1–V1–N1 94.74(11), V1–O1–C1 124.4(2), V1–O2–C19 121.4(2), V1–N1–C50 170.5(3). Symmetry operation used to generate equivalent atoms: i = −x, −y, −z. | ||
51V NMR data for 1–5 and I and II are presented in Table 2, and all vanadyl complexes appear in the range δ −410 to −498 ppm, with the 5-coordinate VO4 centre in II slightly upfield of the other 4-coordinate VO3 containing species. The imido complex 5 appears somewhat downfield, a position which also reflects the presence of the chloride ligand; line widths are also increased in the presence of imido groups.15
Ring opening polymerisation (ROP) studies
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for [CL]:[cat] was best both in the presence or absence of BnOH, over a period of 24 h at 80 °C. For all catalyst systems, runs conducted at temperatures of ≤45 °C or for ≤12 h led to either no polymer or low yields (see Table 3). All systems were relatively well behaved with only one run (run 3) affording a PDI of over 1.80, whilst the majority of runs were below 1.40. The presence of BnOH was also examined for 4 (runs 25–27), and under the optimized conditions, the conversion was about 10% lower whilst the observed Mn was about 30% lower. For 5, bearing a terminal chloride ligand, the use of BnOH was beneficial in terms of % conversion, and the observed molecular weights (Mn) were also higher. In terms of pro-catalyst structure, there appeared to be no advantage in having two metals present given % conversion for 1 ≈ I and II under optimized conditions (runs 3, 6 and 9). In the case of the tetra-phenolate systems, it appears that use of systems (3 and 4) derived from the meta pro-ligand set m-L2mH4 are more effective than those (2) derived from the para pro-ligand p-L2pH4 (runs 13–15 and 16–26 versus 10–12) Table 3. This suggests in 3 and 4 that there is a favourable V⋯V separation, which may favour the coordination of a single monomer to both catalytic centres of the same complex. One centre can then be used as a Lewis acid and the other using its V-OR functionality to attack the carbonyl group. The chloride complex 5, in the presence of BnOH, afforded the highest yield (85%).
:1 for [CL]:[cat] was best both in the presence or absence of BnOH, over a period of 24 h at 80 °C. For all catalyst systems, runs conducted at temperatures of ≤45 °C or for ≤12 h led to either no polymer or low yields (see Table 3). All systems were relatively well behaved with only one run (run 3) affording a PDI of over 1.80, whilst the majority of runs were below 1.40. The presence of BnOH was also examined for 4 (runs 25–27), and under the optimized conditions, the conversion was about 10% lower whilst the observed Mn was about 30% lower. For 5, bearing a terminal chloride ligand, the use of BnOH was beneficial in terms of % conversion, and the observed molecular weights (Mn) were also higher. In terms of pro-catalyst structure, there appeared to be no advantage in having two metals present given % conversion for 1 ≈ I and II under optimized conditions (runs 3, 6 and 9). In the case of the tetra-phenolate systems, it appears that use of systems (3 and 4) derived from the meta pro-ligand set m-L2mH4 are more effective than those (2) derived from the para pro-ligand p-L2pH4 (runs 13–15 and 16–26 versus 10–12) Table 3. This suggests in 3 and 4 that there is a favourable V⋯V separation, which may favour the coordination of a single monomer to both catalytic centres of the same complex. One centre can then be used as a Lewis acid and the other using its V-OR functionality to attack the carbonyl group. The chloride complex 5, in the presence of BnOH, afforded the highest yield (85%).
| Runa | Cat | Temp/°C | Time/h | [CL]0:[Cat]0:[BnOH]0 |
Conv.b (%) | M n,GPC c | M n,cal d | PDIe |
|---|---|---|---|---|---|---|---|---|
| a All reactions were carried out in toluene under nitrogen. b Determined by 1H NMR. c M n values were determined by GPC in THF vs. PS standards and were corrected with a Mark–Houwink factor of 0.56. d Calculated by (F.W. monomer × [monomer]/[cat]) × conversion + F.W. BnOH. e (Mw/Mn) were determined by GPC. | ||||||||
| 1 | 1 | 45 | 24 | 200:1:0 |
84 | 2440 | 19400 |
1.11 |
| 2 | 1 | 60 | 24 | 200:1:0 |
99 | 4450 | 22600 |
1.43 |
| 3 | 1 | 80 | 24 | 200:1:0 |
99 | 5430 | 22600 |
2.01 |
| 4 | I | 45 | 24 | 200:1:0 |
98 | 4600 | 22370 |
1.15 |
| 5 | I | 60 | 24 | 200:1:0 |
99 | 5760 | 22600 |
1.80 |
| 6 | I | 80 | 24 | 200:1:0 |
98 | 5110 | 25600 |
1.66 |
| 7 | II | 45 | 24 | 200:1:0 |
98 | 2860 | 22370 |
1.20 |
| 8 | II | 60 | 24 | 200:1:0 |
98 | 3040 | 22370 |
1.14 |
| 9 | II | 80 | 24 | 200:1:0 |
99 | 5950 | 22600 |
1.32 |
| 10 | 2 | 45 | 24 | 200:1:0 |
55 | 1620 | 12560 |
1.17 |
| 11 | 2 | 60 | 24 | 200:1:0 |
95 | 2850 | 21830 |
1.20 |
| 12 | 2 | 80 | 24 | 200:1:0 |
99 | 4110 | 22600 |
1.14 |
| 13 | 3 | 45 | 24 | 200:1:0 |
— | — | — | — |
| 14 | 3 | 60 | 24 | 200:1:0 |
49 | 1610 | 11410 |
1.15 |
| 15 | 3 | 80 | 24 | 200:1:0 |
99 | 2880 | 22600 |
1.35 |
| 16 | 4 | 80 | 1 | 200:1:0 |
— | — | — | — |
| 17 | 4 | 80 | 6 | 200:1:0 |
49 | 1120 | 11410 |
1.12 |
| 18 | 4 | 80 | 12 | 200:1:0 |
89 | 2500 | 20320 |
1.23 |
| 19 | 4 | 45 | 24 | 200:1:0 |
— | — | — | — |
| 20 | 4 | 60 | 24 | 200:1:0 |
50 | 1620 | 11560 |
1.12 |
| 21 | 4 | 80 | 24 | 100:1:0 |
89 | 2140 | 10270 |
1.16 |
| 22 | 4 | 80 | 24 | 200:1:0 |
99 | 3520 | 22740 |
1.33 |
| 23 | 4 | 80 | 24 | 400:1:0 |
99 | 4920 | 45200 |
1.42 |
| 24 | 4 | 80 | 24 | 600:1:0 |
99 | 5720 | 67800 |
1.47 |
| 25 | 4 | 45 | 24 | 200:2:1 |
10 | 800 | 2280 | 1.13 |
| 26 | 4 | 60 | 24 | 200:2:1 |
97 | 1200 | 22250 |
1.29 |
| 27 | 4 | 80 | 24 | 200:2:1 |
99 | 2510 | 22710 |
1.30 |
| 28 | 5 | 45 | 24 | 200:1:0 |
— | — | — | — |
| 29 | 5 | 60 | 24 | 200:1:0 |
97 | 2170 | 22370 |
1.16 |
| 30 | 5 | 80 | 24 | 200:1:0 |
99 | 3150 | 22600 |
1.21 |
| 31 | 5 | 45 | 24 | 200:2:1 |
— | 440 | — | 1.16 |
| 32 | 5 | 60 | 24 | 200:2:1 |
99 | 3260 | 22710 |
1.14 |
| 33 | 5 | 80 | 24 | 200:2:1 |
99 | 5780 | 22710 |
1.43 |
In the 1H NMR spectra of the resulting PCL (e.g. see ESI, Fig. S7†), for runs involving pro-catalysts with a V-OR moiety present, signals were assignable to a hydroxyl end group (CH2OH) and an alkyl ester (e.g., isopropyl ester for 3). This indicated that the polymerization procedure involved rupture of the monomer acyl–oxygen bond and insertion in the alkoxide–vanadium bond. For runs conducted in the presence of BnOH, 1H NMR spectra were more complicated in terms of end group, with both OBn and OR (e.g. t-Bu for 4) present (see ESI, Fig. S8†).
Interestingly, in the absence of solvent, these systems performed far better at 80 °C, with conversions ≥ 95%, polydispersities ≤ 1.62 (see Table 4) and in general afforded higher observed molecular weight (Mn) polymers (see Table 4). The monomeric tert-butoxide complex 1 was found to afford the best yield (90%) and highest molecular weight (Mn) PCL (∼16300). By contrast, III afforded lowest conversion and highest PDI, which we assume is due to the lack of a readily accessible alkoxide bond (a phenoxide linkage of the tri-phenolate would need to be broken). Interestingly, the isopropoxides 2 and 3 gave very similar results, whilst the tert-butoxide 4 afforded a polymer of much lower molecular weight (Mn). For complex 5, it was necessary to add an equivalent of BnOH to achieve ROP activity (run 7 vs. 8, Table 4), and the resulting polymer was of higher molecular weight (Mn) ∼14000 g mol−1.
| Run | Cat | Time/min | [CL]0:[Cat]0:[BnOH]0 |
M n,GPC a | M n,cal b | PDIc | Conv. (%) |
|---|---|---|---|---|---|---|---|
| a M n values were determined by GPC in THF vs. PS standards and were corrected with a Mark–Houwink factor of 0.56. b (F.W. monomer × [monomer]/[cat]) × conversion + F.W. BnOH. c M w/Mn were determined by GPC. | |||||||
| 1 | 1 | 20 | 200:1:0 |
16270 |
22600 |
1.45 | 99 |
| 2 | I | 20 | 200:1:0 |
4930 | 22600 |
1.26 | 99 |
| 3 | II | 30 | 200:1:0 |
11740 |
21690 |
1.62 | 95 |
| 4 | 2 | 30 | 200:1:0 |
7200 | 22140 |
1.34 | 97 |
| 5 | 3 | 30 | 200:1:0 |
7130 | 21910 |
1.50 | 96 |
| 6 | 4 | 40 | 200:1:0 |
2660 | 22370 |
1.20 | 98 |
| 7 | 5 | 30 | 200:1:1 |
14050 |
22250 |
1.49 | 97 |
| 8 | 5 | 60 | 200:1:0 |
— | — | — | — |
In general for the CL runs, despite the narrow polydispersity, the polymer molecular weights (Mn) were much lower than expected, indicating in all cases that significant trans-esterification reactions were occurring. Further evidence was provided by the MALDI-ToF mass spectra where, as well as the major population of peaks, there was evidence of a second, albeit minor, population (see ESI, Fig S9–S12† in toluene; Fig. S13 and S14,† no solvent).
For 6, a plot of average molecular weight (Mn) versus against conversion (Fig. 7, runs 21–24 Table 3) exhibited a linear relationship. Given the plot also shows that the PDI remained narrow, it suggests that under these conditions the ROP by 6 is proceeding in a living manner.
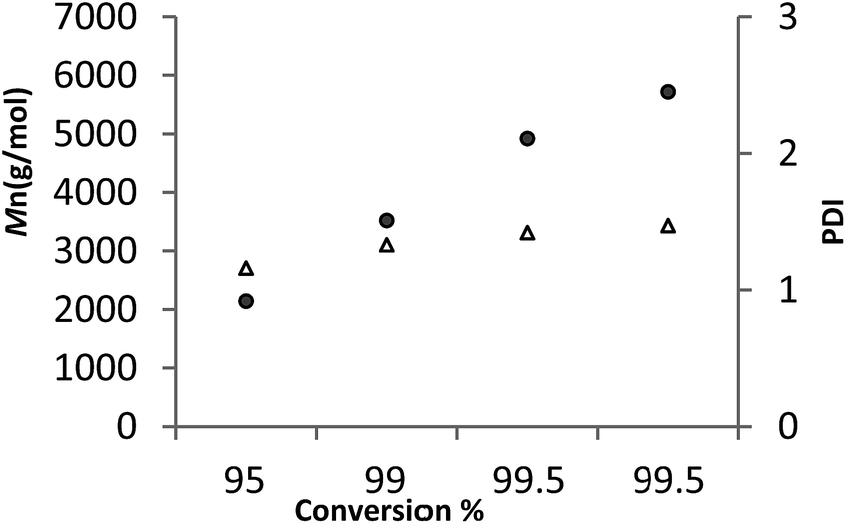 | ||
| Fig. 7 M n (•) and Mw/Mn (Δ) vs. monomer conversion in the ROP of ε-CL. | ||
The production of only low molecular weight polymers using alkoxy vanadium systems has been noted previously.9 Herein, there was little correlation of 51V NMR signal (Table 2) versus catalytic activity (see ESI, Fig. S15†).
:1 for L-LA to pro-catalyst, it was found that at temperatures below 80 °C, there was no catalytic activity even after 35 h. At 80 °C, there was no activity after 6 h, and polymer was only isolated at 24 h affording a yield of 20%. Prolonging the reaction time increased the yield to 50%. Further increasing the temperature to 110 °C afforded only a slight improvement in yield, with a slight increase in PDI. Varying the ratio of L-LA to pro-catalyst led to a slight improvement in the yield (55%), together with an increase in the molecular weight (Mn) and a slight broadening of the PDI. Given these results, the other complexes were screened using a ratio of 200:1 for L-LA to pro-catalyst, and in the case of 5, screening was conducted both in the absence and presence of BnOH (see Table 5).
| Runa | Cat | Temp/°C | Time/h | [LA]0:[Cat]0:[BnOH]0 |
Conv.b (%) | M n,GPC c | M n,cal d | PDIe |
|---|---|---|---|---|---|---|---|---|
| a All reactions were carried out in toluene under nitrogen. b Determined by 1H NMR. c M n values were determined by GPC in THF vs. PS standards and were corrected with a Mark–Houwink factor of 0.58. d (F.W. monomer × [monomer]/[cat]) × conversion + F.W. BnOH. e (Mw/Mn) were determined by GPC. | ||||||||
| 1 | 1 | 45 | 31 | 200:1:0 |
— | — | — | — |
| 2 | 1 | 60 | 24 | 200:1:0 |
10 | 1480 | 2880 | 1.57 |
| 3 | 1 | 80 | 6 | 200:1:0 |
— | — | — | — |
| 4 | 1 | 80 | 24 | 200:1:0 |
54 | 2310 | 15570 |
1.24 |
| 5 | 1 | 80 | 35 | 200:1:0 |
50 | 2100 | 14410 |
1.26 |
| 6 | I | 45 | 34 | 200:1:0 |
— | — | — | — |
| 7 | I | 60 | 34 | 200:1:0 |
— | — | — | — |
| 8 | I | 80 | 6 | 200:1:0 |
— | — | — | — |
| 9 | I | 80 | 24 | 200:1:0 |
54 | 2930 | 15850 |
1.73 |
| 10 | I | 80 | 31 | 200:1:0 |
54 | 2640 | 15850 |
1.16 |
| 11 | II | 60 | 34 | 200:1:0 |
— | — | — | — |
| 12 | II | 80 | 6 | 200:1:0 |
— | — | — | — |
| 13 | II | 80 | 24 | 200:1:0 |
58 | 3010 | 16720 |
1.46 |
| 14 | II | 80 | 34 | 200:1:0 |
60 | 3440 | 17300 |
1.31 |
| 15 | 2 | 60 | 31 | 200:1:0 |
— | — | — | — |
| 16 | 2 | 80 | 6 | 200:1:0 |
— | — | — | — |
| 17 | 2 | 80 | 24 | 200:1:0 |
50 | 2120 | 14410 |
1.34 |
| 18 | 2 | 80 | 36 | 200:1:0 |
58 | 2190 | 16720 |
1.30 |
| 19 | 3 | 45 | 33 | 200:1:0 |
— | — | — | — |
| 20 | 3 | 60 | 35 | 200:1:0 |
— | — | — | — |
| 21 | 3 | 80 | 6 | 200:1:0 |
— | — | — | — |
| 22 | 3 | 80 | 24 | 200:1:0 |
15 | 1340 | 4320 | 1.11 |
| 23 | 3 | 80 | 36 | 100:1:0 |
52 | 2150 | 7490 | 1.11 |
| 24 | 3 | 80 | 36 | 200:1:0 |
52 | 2510 | 14990 |
1.09 |
| 25 | 3 | 80 | 36 | 400:1:0 |
60 | 34590 |
34590 |
1.26 |
| 26 | 3 | 80 | 36 | 600:1:0 |
62 | 3760 | 53620 |
1.29 |
| 27 | 3 | 110 | 31 | 200:1:0 |
51 | 2010 | 14700 |
1.26 |
| 28 | 4 | 60 | 30 | 200:1:0 |
9 | 870 | 2590 | 1.23 |
| 29 | 4 | 80 | 6 | 200:1:0 |
— | — | — | — |
| 30 | 4 | 80 | 24 | 200:1:0 |
20 | 1930 | 5760 | 1.01 |
| 31 | 4 | 80 | 31 | 200:1:0 |
45 | 2460 | 12970 |
1.10 |
| 32 | 5 | 80 | 31 | 200:1:0 |
— | — | — | — |
| 33 | 5 | 80 | 6 | 200:1:1 |
— | — | — | — |
| 34 | 5 | 80 | 24 | 200:1:1 |
53 | 3400 | 15380 |
1.29 |
| 35 | 5 | 110 | 31 | 200:1:1 |
55 | 3640 | 15960 |
1.35 |
In the case of 4, differing from 3 only in the nature of the alkoxide (tert-butoxide versus isopropoxide) there was some activity at 60 °C over 30 h, though the yield was low (10%). Reactions conducted at 80 °C afforded yields slightly lower than observed for 3; molecular weights (Mn) were similar. In the case of complex 2, which differs from 3 in the nature of the tetra-phenolate employed (i.e. para versus meta), activity was observed at 80 °C on prolonging the reaction time. Yields using 2 after 24 h were typically higher than for 3, but then after 36 h, the yields were approximately the same (slightly higher using 3); molecular weights (Mn) followed the same trend. PDIs for runs employing 2 were higher than those for 3. However, these results, unlike those for ε-caprolactone, did not suggest that use of the meta ligand had any beneficial effect in terms of the distance between the two vanadium centres and the resultant ROP activity. In the case of the imido complex 5 (a chloride complex), it proved necessary here to add BnOH to afford an active system. We note however that chlorides have previously been shown to be capable of the ROP of lactide.16 At 80 °C, activity was observed after 24 h, with yields similar to the vanadyl complex 2, but with higher molecular weight (Mn) polymers formed.
Comparing results for 1versusI suggests that there is no benefit in having two vanadyl centers present rather than one. Indeed, results for 1 suggest the opposite given that 1 can operate at 60 °C and also affords superior yields at 80 °C. Results for III are similar to those of II.
In all cases, observed molecular weights (Mn) are far lower than calculated values. In contrast to the ε-caprolactone screening, conducting the L-LA ROP runs in the absence of any solvent did not afford improved results and actually afforded little or no polymer.
As for PCL, the observed molecular weights (Mn) for the PLA are lower than the calculated values, and in the MALDI-ToF spectra (see ESI, Fig. S16–S18†) there was evidence of a second population consistent with some transesterification processes occurring.
For 3, a plot (Fig. 8, runs 23–26, Table 5) of the average molecular weight (Mn) of the poly(L-LA) as a function of the monomer conversion was linear, and with consistently low PDI values suggestive of a living process.
 | ||
| Fig. 8 M n (Δ) and Mw/Mn (•) vs. monomer conversion in the ROP of L-LA. | ||
| Runa | Cat | Temp/°C | Time/h | [LA]0:[Cat]0:[BnOH]0 |
Conv.b (%) | M n,GPC c | M n,cal d | PDIe | Prf |
|---|---|---|---|---|---|---|---|---|---|
| a All reactions were carried out in toluene under nitrogen. b Determined by 1H NMR. c M n values were determined by GPC in THF vs. PS standards and were corrected with a Mark–Houwink factor of 0.58. d Calculated from (F.W. monomer × [monomer]/[cat]) × conversion + F.W. BnOH. e M w/Mn were determined by GPC. f Determined by analysis of the tetrad signal in the 1H NMR spectrum. | |||||||||
| 1 | 1 | 80 | 24 | 200:1:0 |
80 | 6050 | 23060 |
1.30 | 0.61 |
| 2 | I | 60 | 24 | 200:1:0 |
— | — | — | — | — |
| 3 | I | 80 | 24 | 200:1:0 |
70 | 3650 | 20180 |
1.15 | 0.60 |
| 4 | II | 60 | 24 | 200:1:0 |
— | — | — | — | — |
| 5 | II | 80 | 24 | 200:1:0 |
78 | 3860 | 22480 |
1.09 | 0.63 |
| 6 | 2 | 60 | 24 | 200:1:0 |
— | — | — | — | — |
| 7 | 2 | 80 | 24 | 100:1:0 |
65 | 1460 | 9370 | 1.13 | — |
| 8 | 2 | 80 | 24 | 200:1:0 |
80 | 2720 | 23060 |
1.19 | 0.58 |
| 9 | 2 | 80 | 24 | 400:1:0 |
75 | 3450 | 43240 |
1.24 | 0.58 |
| 10 | 2 | 80 | 24 | 600:1:0 |
75 | 3570 | 64860 |
1.23 | 0.58 |
| 11 | 2 | 110 | 24 | 200:1:0 |
75 | 2440 | 21620 |
1.21 | 0.58 |
| 12 | 3 | 60 | 24 | 200:1:0 |
— | — | — | — | — |
| 13 | 3 | 80 | 12 | 200:1:0 |
44 | 790 | 12970 |
1.17 | — |
| 14 | 3 | 80 | 24 | 200:1:0 |
83 | 2860 | 25940 |
1.18 | 0.58 |
| 15 | 3 | 110 | 24 | 200:1:0 |
75 | 2480 | 21620 |
1.27 | 0.61 |
| 16 | 4 | 80 | 24 | 200:1:0 |
79 | 2120 | 23060 |
1.25 | 0.61 |
| 17 | 4 | 110 | 24 | 200:1:0 |
75 | 4120 | 21620 |
1.23 | 0.61 |
| 18 | 5 | 80 | 24 | 200:1:1 |
— | — | — | — | — |
| 19 | 5 | 110 | 24 | 200:1:1 |
78 | 2210 | 22590 |
1.10 | 0.58 |
| 20 | 5 | 130 | 24 | 200:1:0 |
— | — | — | — | — |
 | ||
| Fig. 9 M n (Δ) and Mw/Mn (•) vs. monomer conversion in the ROP of rac-LA. | ||
Observed molecular weights (Mn) were again lower than calculated values, and MALDI-Tof spectra (e.g. Fig. S19, ESI†) also revealed a number of minor populations.
As for L-lactide, use of no solvent afforded little or no observed catalytic activity.
To assign the stereochemistry of the PLA polymers we employed 2D J-resolved 1H NMR spectroscopy and assigned the peaks by reference to the literature.17 Representative spectra for runs 1, 5 and 17 are given in the ESI (Fig. S20–S22†), with the assignments given on the respective figures. These systems gave moderately isotactic PLA with a Pr value in the range 0.58–0.63.
Runs conducted in different solvents, namely THF and CH2Cl2 resulted in little or no polymer.
The presence of cyclic PLA was ruled out by comparison with literature MALDI-ToF and 1H NMR spectra.18
:200:1 for CL:LA:pro-cat over 24 h (×2). In all cases (Table 7), good yields (65–83%) of co-polymer were formed, but with low lactide content (2.5–9.0%) as observed from 1H NMR spectra (ESI, Fig. S23†); the highest % incorporation of LA (9%) was found for 7 in the presence of BnOH. In the 1H NMR, the end groups for alkoxide and hydroxyl were also evident. Observed molecular weights were in general higher than those observed for the homo-polymerisations conducted in toluene. Thermal analysis of the co-polymer by DSC revealed two melting points at 55.1 °C (PCL) and 170.5 °C (PLA), see ESI Fig. S24.† If the addition of the monomers was reversed, i.e.L-LA added first, no co-polymer was isolated after work-up. This suggested that a PLA chain end was not capable of PCL chain growth.
| Runa | Complex | CL:LAb |
Yield% | M n c | M w/Mnd |
|---|---|---|---|---|---|
| a All reactions were carried out in toluene under nitrogen under optimum condition 24 h CL/24 h L-LA (80 °C). b Ratio of LA to CL observed in the co-polymer by 1H NMR. c M n values were determined by GPC in THF vs. PS standards and were corrected with a Mark–Houwink factor (Mn,GPC × 0.56 × % PCL + Mn,GPC × 0.58 × % PLLA). d PDI were determined by GPC. | |||||
| 1 | 1 | 390:10 |
83 | 15740 |
1.87 |
| 2 | I | 383:17 |
77 | 3840 | 1.24 |
| 3 | II | 379:21 |
68 | 3600 | 1.16 |
| 4 | 2 | 385:15 |
73 | 4890 | 1.54 |
| 5 | 3 | 383:17 |
71 | 4560 | 1.66 |
| 6 | 4 | 380:20 |
66 | 5790 | 1.43 |
| 7 | 5 | 364:46 |
65 | 3400 | 1.26 |
| Runa | Complex | CL:LAb |
Yield% | M n c | M w/Mnd |
|---|---|---|---|---|---|
| a All reactions were carried out in toluene under nitrogen under optimum condition 24 h CL/24 h rac-LA (80 °C). b Ratio of LA to CL observed in the co-polymer by 1H NMR spectroscopy. c M n values were determined by GPC in THF vs. PS standards and were corrected with a Mark–Houwink factor (Mn,GPC × 0.56 × % PCL + Mn,GPC × 0.58 × % PLLA). d PDI were determined by GPC. | |||||
| 1 | 1 | 382:18 |
88 | 5850 | 1.42 |
| 2 | I | 375:25 |
73 | 3210 | 1.92 |
| 3 | II | 363:73 |
70 | 3910 | 1.21 |
| 4 | 2 | 360:40 |
75 | 3260 | 1.71 |
| 5 | 3 | 375:25 |
68 | 3840 | 1.60 |
| 6 | 4 | 367:33 |
60 | 3270 | 1.46 |
| 7 | 5 | 343:57 |
70 | 1770 | 1.18 |
Given the low molecular weight products isolated during these studies, the presence of impurities (e.g. lactic acid from non-recrystallized rac-lactide) acting as chain transfer agents (or co-initiator) cannot be ruled out. However, we note that there is interest in low molecular weight poly(lactide/caprolactone) polymers as bio adhesives.19
Conclusions
In conclusion, we have examined the ROP behaviour of a series of vanadyl complexes bearing chelating di-, tri- and tetra-phenolate ligands towards ε-caprolactone, L-lactide or rac-lactide with and without solvent present, and the co-polymerisation of ε-CL with lactide. For the homo-polymerisation of ε-CL, under the optimized conditions in toluene, yields were typically of the order of 70%. It was observed that there was no advantage in having two metals present (cf. to one), whilst for the tetra-phenolates use of the meta ligand set appeared beneficial (cf. the para ligand set), which perhaps reflects the closer proximity of the metal centres in the former. Conducting the runs in the absence of solvent led to higher conversions, typically >95%. For the ROP of L-lactide, the performances of 1–5 (and I and II) were not so good, affording PLA in only low to moderate (≤55%) yields at higher temperatures and over prolonged reaction periods. There was no evidence of any beneficial cooperative effects and conducting the runs in the absence of solvent afforded no improvement. Results for rac-lacide were similar to L-lactide; 2D J-resolved 1H NMR spectroscopy indicated the formation of moderately isotactic PLA. Co-polymerisation of CL with L-lactide afforded good yields (65–83%) of co-polymer, but with low lactide content (2.5–9.0%); reversing the order of monomer addition resulted in no product. Similar results were observed for co-polymerisation of CL with rac-lactide.Experimental
General
All manipulations were carried out under an atmosphere of dry nitrogen using conventional Schlenk and cannula techniques or in a nitrogen-filled glove box. Diethyl ether and tetrahydrofuran were refluxed over sodium and benzophenone. Toluene was refluxed over sodium. Dichloromethane and acetonitrile were refluxed over calcium hydride. All solvents were distilled and degassed prior to use. IR spectra (nujol mulls, KBr or NaCl windows) were recorded on a Nicolet Avatar 360 FT IR spectrometer; 1H NMR spectra were recorded at room temperature on a Varian VXR 400 S spectrometer at 400 MHz or a Gemini 300 NMR spectrometer or a Bruker Advance DPX-300 spectrometer at 300 MHz. The 1H NMR spectra were calibrated against the residual protio impurity of the deuterated solvent. Elemental analyses were performed by the elemental analysis service at the London Metropolitan University or the University of Hull. Matrix Assisted Laser Desorption/Ionization-Time of Flight (MALDI-ToF) mass spectrometry was performed on Bruker autoflex III smart beam in linear mode. MALDI-ToF mass spectra were acquired by averaging at least 100 laser shots. 2,5-Dihydroxylbenzoic acid was used as matrix and tetrahydrofuran as solvent. Sodium chloride was dissolved in methanol and used as the ionizing agent. Samples were prepared by mixing 20 μl of polymer solution in tetrahydrofuran (2 mg ml−1) with 20 μl of matrix solution (10 mg ml−1) and 1 μl of a solution of ionizing agent (1 mg ml−1). Then 1 ml of these mixtures was deposited on a target plate and allowed to dry in air at room temperature. L-Lactide (99.7%, with D-lactide content less than 1%) was provided by Shenzhen Brightchina Industrial Co. Ltd (Shenzhen, China) as monomer. rac-Lactide and ε-caprolactone were purchased from Sigma Aldrich and were used as received. Benzyl alcohol was distilled over CaH2 under vacuum. Other solvents were all of analytical grade and provided by Kelong Chemical Reagent Factory of Chengdu (Chengdu, China), and were used without further purification unless noted. Complexes I and II were prepared as reported in the literature.11Ring opening polymerisation procedure
Polymerisation reactions were performed in a Schlenk tube equipped with a magnetic stirrer. For solution polymerisation, a mixture of monomer (14 mmol of ε-caprolactone, L-lactide or rac-lactide), vanadyl complexes 1–5 or I and II (0.14 mmol) and BnOH (0.14 mmol, if needed) were added into a Schlenk tube at room temperature under nitrogen protection. After 3 cycles of gassing and degassing, pre-degassed toluene (7 ml) was added to the reaction mixture under nitrogen protection. The polymerisation was started by placing the reaction mixture into an oil bath pre-heated to the polymerisation temperature (Tp). For bulk polymerisation, all reaction conditions were the same except that no solvent was used. In all cases, the polymerisation was stopped by addition of 1 ml methanol after the prescribed reaction time. Products with different polymerisation times were taken out with a syringe and precipitated into cold methanol with magnetic stirring to eliminate residual catalyst. Precipitates were filtrated, washed with methanol and dried before GPC determination.Co-polymerisations were conducted by adding monomer 1 (3.200 mmol) to the catalyst (0.016 mmol) in toluene pre-heated to 80 °C and stirring for 24 h, and then adding monomer 2 (3.320 mmol) and stirring for a further 24 h. The polymerisation mixture was quenched by adding methanol (1.0 ml), and the resultant solution was then poured into methanol (200 ml), the precipitate collected and dried in vacuo.
Crystallography
Single crystal diffraction data were collected by the UK National Crystallography Service using a Rigaku FR-E+ diffractometer. This operates with a SuperBright rotating anode X-ray generator and high flux optics. This is designed to deal with the most challenging samples sent to the service.Despite the high flux, the crystal of 1 examined was found to scatter X-rays very poorly and little appreciable diffraction was observed beyond ∼1.1 Å. It was possible to solve the structure using this data and routine refinements of a structural model were possible. It was possible to use anisotropic displacement parameters for all non-hydrogen atoms and the refinement was stable with no unusual features. Although the crystal examined was weakly scattering the solution is sound and it gives extremely useful chemical information.
The data for 2, although weak, are more routine and standard procedures were applied in structure solution.
Structures were solved using Direct Methods implemented within SHELXS-2013 and refined within SHELXL-2014.20
Further details are provided in Table S2.†
Acknowledgements
LJ thanks the National Natural Science Foundation of China (No. 50873067 and No. 51403140) and Research Fund for the Doctoral Program of Higher Education of China (No. 20110181110032 and International Scientific and Technological Cooperation Projects (No. 2010DFA54460) of China) for financial support. CR thanks the EPSRC for the award of a travel grant and Astatech Ltd (Chengdu) for loan of [VO(OnPr)3] and di-phenol.Notes and references
- For recent reviews see: (a) A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411 CrossRef CAS PubMed; (b) C. K. Williams and M. A. Hillmyer, Polym. Rev., 2008, 48, 1 CrossRef CAS; (c) E. S. Place, J. H. George, C. K. Williams and M. H. Stevens, Coord. Chem. Rev., 2009, 38, 1139 CAS.
- (a) M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484 RSC; (b) A. Arbaoui and C. Redshaw, Polym. Chem., 2010, 1, 801 RSC; (c) X. Rong and C. Chunxia, Progr. Chem., 2012, 24, 1519 Search PubMed.
- R. Mazarro, I. Gracia, J. F. Rodriquez, G. Storti and M. Morbidelli, Polym. Int., 2012, 61, 265 CrossRef CAS.
- (a) C. Redshaw, Dalton Trans., 2010, 39, 5595 RSC; (b) K. Nomura and W. Zhang, Chem. Rev., 2011, 111, 2342 CrossRef CAS PubMed; (c) J.-Q. Wu and Y.-S. Li, Coord. Chem. Rev., 2011, 255, 2303 CrossRef CAS; (d) C. Redshaw, M. J. Walton, M. R. J. Elsegood, T. J. Prior and K. Michiue, RSC Adv., 2015, 5, 89783 RSC.
- C. Redshaw, M. R. J. Elsegood, J. A. Wright, H. Baillie-Johnson, T. Yamato, S. de Giovanni and A. Mueller, Chem. Commun., 2012, 48, 1129 RSC.
- (a) Y. Kim, P. N. Kapoor and J. G. Verkade, Inorg. Chem., 2002, 41, 4834 CrossRef CAS PubMed; (b) C. Alonso-Moreno, A. Antiňolo, J. C. Garcia-Martinez, S. Garcia-Yuste, I. López-Solera, A. Otero, J. C. Perez-Flores and M. T. Tercero-Morales, Eur. J. Inorg. Chem., 2012, 1139 CrossRef CAS; (c) T. K. Saha, M. Mandel, M. Thunga, D. Chakraborty and V. Ramkumar, Dalton Trans., 2012, 42, 10304 RSC; (d) Y. Al-Khafaji, X. Sun, T. J. Prior, M. R. J. Elsegood and C. Redshaw, Dalton Trans., 2015, 44, 12349 RSC.
- Use of di-phenols: (a) B.-T. Ko and C.-C. Lin, Macromolecules, 1999, 32, 8296 CrossRef CAS; (b) I. Taden, H.-C. Kang, W. Massa, T. P. Spaniol and J. Okuda, Eur. J. Inorg. Chem., 2000, 441 CrossRef CAS; (c) H.-L. Chen, B.-T. Ko, B.-H. Huang and C.-C. Lin, Organometallics, 2001, 20, 5076 CrossRef CAS; (d) B. T. Ko and C.-C. Lin, J. Am. Chem. Soc., 2001, 123, 7973 CrossRef CAS PubMed; (e) Y.-C. Liu, B.-T. Ko and C.-C. Lin, Macromolecules, 2001, 34, 6196 CrossRef CAS; (f) M.-L. Hsueh, B.-H. Huang and C.-C. Lin, Macromolecules, 2002, 35, 5763 CrossRef CAS; (g) M. H. Chisholm, C.-C. Lin, J. C. Gallucci and B.-T. Ko, Dalton Trans., 2003, 406 RSC; (h) T.-C. Liao, Y.-L. Huang, B.-H. Huang and C.-C. Lin, Macromol. Chem. Phys., 2003, 204, 885 CrossRef CAS; (i) Y. Takashima, Y. Nakayama, T. Hirao, H. Yasuda and A. Harada, J. Organomet. Chem., 2004, 689, 612 CrossRef CAS; (j) M.-L. Shueh, Y.-S. Wang, B.-H. Huang, C.-Y. Kuo and C.-C. Lin, Macromolecules, 2004, 37, 5155 CrossRef CAS; (k) R.-M. Ho, Y.-W. Chiang, C.-C. Tsai, C.-C. Lin, B.-T. Ko and B.-H. Huang, J. Am. Chem. Soc., 2004, 126, 2704 CrossRef CAS PubMed; (l) T.-L. Yu, C.-C. Wu, C.-C. Chen, B.-H. Huang, J. Wu and C.-C. Lin, Polymer, 2005, 46, 5909 CrossRef CAS; (m) Y. Yao, X. Xu, B. Liu, Y. Zhang, Q. Shen and W.-T. Wong, Inorg. Chem., 2005, 44, 5133 CrossRef CAS PubMed; (n) X. Xu, Z. Zhang, Y. Yao, Y. Zhang and Q. Shen, Inorg. Chem., 2007, 46, 9379 CrossRef CAS PubMed; (o) M.-L. Hsueh, J. Wu and C.-C. Lin, Macromolecules, 2005, 38, 9482 CrossRef CAS; (p) J. Wu, Y.-Z. Chen, W.-C. Hung and C.-C. Lin, Organometallics, 2008, 27, 4970 CrossRef CAS; (q) Z. Liang, X. Ni, X. Li and Z. Shen, Inorg. Chem. Commun., 2011, 14, 1948 CrossRef CAS; (r) C.-Y. Li, P.-S. Chen, S.-J. Hsu, C.-H. Lin, H.-Y. Huang and B.-T. Ko, J. Organomet. Chem., 2012, 716, 175 CrossRef CAS; (s) X. Xu, X. Pan, S. Tang, X. Lv, L. Li, J. Wu and X. Zhao, Inorg. Chem. Commun., 2013, 29, 89 CrossRef CAS; (t) Z. Liang, M. Zhang, X. Ni, X. Li and Z. Shen, Inorg. Chem. Commun., 2013, 29, 145 CrossRef CAS.
- Use of tetra-phenols: J. Zhang, C. Jian, Y. Gao, L. Wang, N. Tang and J. Wu, Inorg. Chem., 2012, 51, 13380 CrossRef CAS PubMed.
- (a) C. Redshaw, L. Warford, S. H. Dale and M. R. J. Elsegood, Chem. Commun., 2004, 1954 RSC; (b) D. Homden, C. Redshaw, L. Warford, D. L. Hughes, J. A. Wright, S. H. Dale and M. R. J. Elsegood, Dalton Trans., 2009, 8900 RSC.
- P. J. Toscano, E. J. Schermerhorn, C. Dettelbacher, D. Macherone and J. Zubieta, J. Chem. Soc., Chem. Commun., 1991, 933 RSC.
- A. Arbaoui, D. Homden, C. Redshaw, J. A. Wright, S. H. Dale and M. R. J. Elsegood, Dalton Trans., 2009, 8911 RSC.
- T. Moriuchi, M. Nishina and T. Hirao, Angew. Chem., Int. Ed, 2010, 49, 83 CrossRef CAS PubMed.
- (a) A. L. Spek, Acta Crystallogr., 1990, A46, C34 Search PubMed; (b) P. V. D. Sluis and A. L. Spek, Acta Crystallogr., 1990, A46, 194 CrossRef.
- (a) W. Clegg, M. R. J. Elsegood, S. J. Teat, C. Redshaw and V. C. Gibson, J. Chem. Soc., Dalton Trans., 1998, 3037 RSC; (b) W. Clegg, J. Chem. Soc., Dalton Trans., 2000, 3223 RSC.
- (a) E. A. Maatta, Inorg. Chem., 1984, 23, 2561–2562 CrossRef; (b) D. D. Devore, J. D. Lichtenhan, F. Takusagawa and E. A. Maatta, J. Am. Chem. Soc., 1987, 109, 7408 CrossRef CAS.
- See for example (a) Y. Takashima, Y. Nakayama, K. Watanabe, T. Itono, N. Ueyama, A. Nakamura, H. Yasuda, A. Harada and J. Okuda, Macromolecules, 2002, 35, 7358 CrossRef; (b) Y. Kim, G. K. Jnaneshwara and J. G. Verkade, Inorg. Chem., 2003, 42, 1437 CrossRef CAS PubMed.
- A. Grala, J. Ejfler, L. B. Jerzykiewicz and P. Sobota, Dalton Trans., 2011, 40, 4042 RSC.
- E. Piedra-Arroni, C. Ladavière, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2013, 135, 13305 CrossRef PubMed.
- S. Sriputtirat, W. Boonkong, S. Pengprecha, A. Petsom and N. Thongchul, Adv. Chem. Eng. Sci., 2012, 2, 15 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ORTEPs and X-ray crystallographic files CIF format for the structure determinations of compounds 1, 2, 3·2CH2Cl2, 4·2CH2Cl2, 4·3CH2Cl2 and 5·2CH3CN. CCDC 1040815, 1404533, 1404524–1404526 and 1404529. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra24816g |
| This journal is © The Royal Society of Chemistry 2016 |