
Poly(butylene succinate) bionanocomposites: a novel bio-organo-modified layered double hydroxide for superior mechanical properties†
Abstract
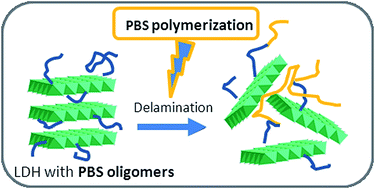
Bionanocomposites based on poly(butylene succinate) and a novel organo-modified layered double hydroxide have been prepared by in situ polymerization. In order to enhance the compatibilization of the inorganic filler with the polymer matrix, an oligomer of PBS was intercalated between the layers of the clay. Composites with different percentages of filler (1, 3, 5, 10 wt%) and two different divalent cations (Zn or Mg) were prepared. The thermal, rheological and mechanical properties of the samples were investigated. The results showed that the materials feature a high thermal stability, more specifically so for the composites containing Zn2+ cations. Rheological investigations highlighted a significant chain extender effect of the filler toward the matrix, thus revealing a huge reinforcing effect imparted by the clays, especially for composites with Mg2+ cations. Such findings were also supported by an increase up to 30% of the tensile and flexural strength for a PBS composite loaded with 3 wt% of Mg/Al-LDH, thus suggesting a uniform level of dispersion and a pronounced interfacial interaction between the filler and polymer.
 Please wait while we load your content...
Please wait while we load your content...

