
Self-ordering dual-layered honeycomb nanotubular titania: a study in formation mechanisms
Abstract
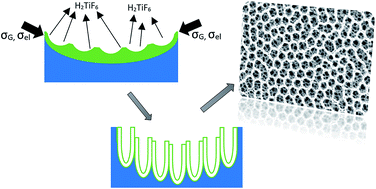
A new method for creating a honeycomb morphology of TiO2 nanotubular arrays has been proposed using a two-step anodization process. The first anodization step utilizes fluoride containing ethylene glycol as an electrolyte (EG-solution). The anodic oxide formed during the first anodization step was removed to expose hemispherical nanoindentations created on the substrate. These nanoimprints acted as templates for honeycomb morphology during the second step of anodization that was carried out in fluoride containing phosphoric acid solution (PA-solution). Several small diameter nanotubes were grown in each honeycomb cell during the second anodization step. The formation of such honeycomb morphology required a threshold concentration of Ti4+ in the anodization electrolyte. Below the threshold Ti4+ concentration (4 × 10−3 M in this investigation), a good honeycomb morphology was not observed. The formation mechanism proposed is based on weakly linear surface perturbation theory aided by compressive growth stresses and a decrease in surface energy due to the presence of [TiF6]2− species.
 Please wait while we load your content...
Please wait while we load your content...

