DOI:
10.1039/C5RA24615F
(Paper)
RSC Adv., 2016,
6, 7712-7716
Synthesis of nitrate ester and nitramine derivatives of polyfluoro alkyl compounds for high energy materials†
Received
20th November 2015
, Accepted 6th January 2016
First published on 11th January 2016
Abstract
Five new poly-fluorine containing molecules were synthesized from economical and commercially available materials for high energy material applications by straightforward routes. All the synthesized molecules were characterized using IR, NMR spectroscopy, elemental analysis, single crystal X-ray diffraction and thermal (TG-DTA), impact, and friction stability measurements. In addition, the crystal structures of 4, 7 and 8 are presented. The synthesized poly-fluorine compounds were found to have reasonable detonation properties based on calculations (EXPLO5 v6.01), which are in the range of TNT and RDX. These compounds are thermally stable (Tdec > 150 °C) and are insensitive toward impact and friction.
Introduction
In recent years, many efforts have been made to find suitable oxidizers and high energy materials (HEMs).1 Many research publications that deal with energetic materials suggest that a better performance of the material is mainly governed by oxygen balance, density, the heat of formation, amounts of gaseous products generated and their burning rate. Considering these observations, energetic materials containing polyfluoroalkyl, nitrate ester (–ONO2) and nitramine (–NHNO2) were designed and synthesized with a view to developing materials with tuned and diverse energetic properties. Earlier, energetic materials containing pentafluorosulfanyl (–SF5),2 trifluoromethyl (–CF3),3 and poly-fluoroalkyl4 were reported to have reasonable densities, reduced sensitivities, and good thermal stabilities. Also, fluorine-containing compounds result in high densities compared to their hydrocarbon analogues due to the large electronegativity of fluorine which gives rise to intra- and inter-molecular interactions. In hydrocarbons, the replacement of hydrogen with fluorine increases the overall oxygen balance which results in formulations with higher performance. Furthermore, the high dielectric constant and electron-withdrawing capability of fluorine-containing groups lead to greater chemical resistance and hydrophobicity.5 Ionic salts which have longer alkyl and polyfluoroalkyl substituents are known to lower melting points due to a less-ordered packing in the solid. Moreover, with an elongation of the fluoroalkyl substituent and increase in the concentration of fluorine, compounds were found to be denser.6 On decomposition, it is expected that fluorine will combine with a hydrogen source and produce a larger number of moles of gaseous products per gram of explosive. A greater energy release can be expected due to the formation of HF in the detonation. The fluorine-containing compositions of Magnesium–Teflon–Viton (MTV) are commonly used in flares, pyrotechnics and defence applications. The dissociation reaction of Teflon and the oxidation of magnesium influence the flame temperature which in turn improves the performance of the compositions. Previously, Shreeve et al. reported the potential applications of fluoro compounds as ionic liquids,7 and energetic materials.8 Christe et al. reported various fluorine-containing compounds and energetic salts.9 Klapötke et al. reported fluorine-containing compounds for application as an oxidizer in propellant formulations.10 Poly-fluoro nitramines and poly-fluoro nitrate esters are expected to have potential HEM applications because of their reasonable detonation properties and lower sensitivity. However, we believe that the syntheses and energetic properties of these compounds have not been explored so far.
Results and discussion
Synthesis
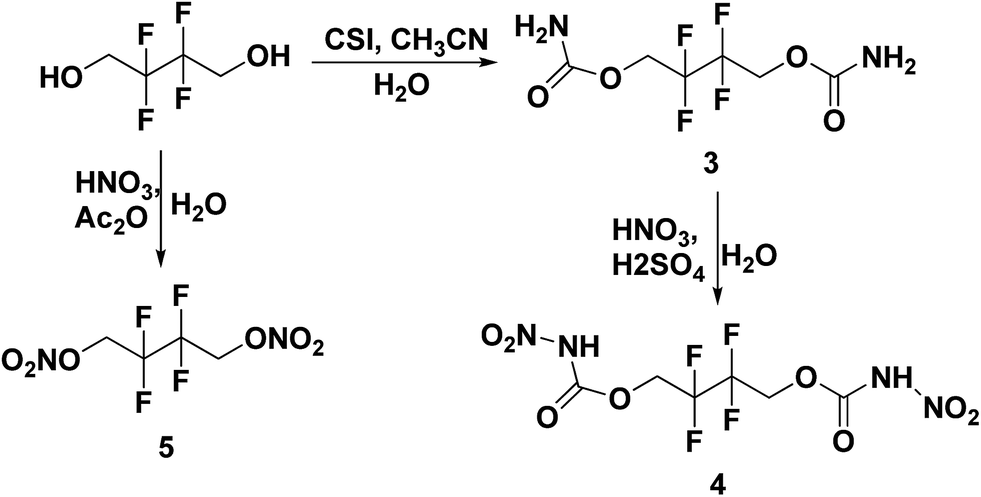
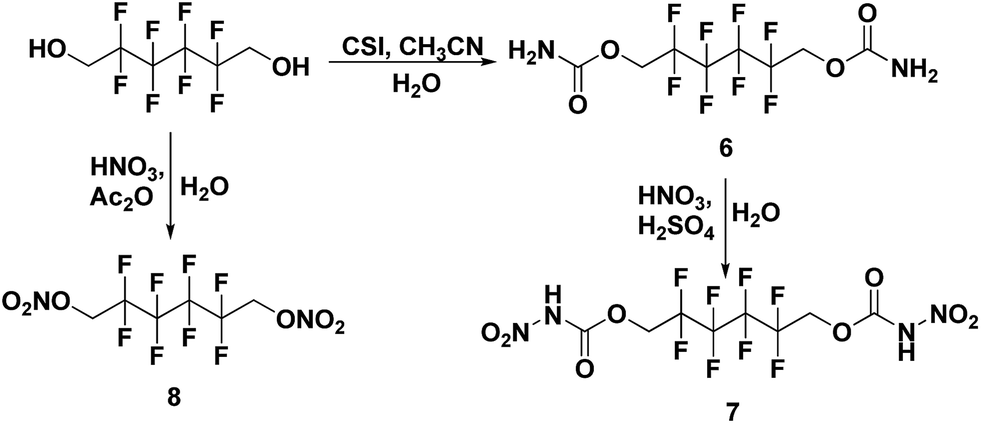
All the syntheses performed in this work start from commercially available starting materials, 2,2,2-trifluoroethanol (in Scheme 1), 2,2,3,3-tetrafluoro-1,4-butanediol (in Scheme 2) and 2,2,3,3,4,4,5,5-octafluoro-1,6-hexanediol (in Scheme 3). 2,2,2-Trifluoroethyl carbamate (1) was synthesized from 2,2,2-trifluoroethanol and chlorosulfonyl isocyanate (CSI) according to the literature-reported procedure.11 Subsequently, 2,2,2-trifluoroethyl carbamate (1) was nitrated to 2,2,2-trifluoroethyl nitrocarbamate (2) using fuming HNO3 and conc. H2SO4. Along similar lines, 2,2,3,3-tetrafluoro-1,4-butanediol treated with CSI to give 2,2,3,3-tetrafluorobutane-1,4-diyl dicarbamate (3) and further nitrated to yield 2,2,3,3-tetrafluorobutane-1,4-diyl bis(nitrocarbamate) (4). Nitration of 2,2,3,3-tetrafluoro-1,4-butanediol was also performed with nitric acid and acetic anhydride, furnishing 2,2,3,3-tetrafluorobutane-1,4-diyl dinitrate (5). The carbamate formation and nitration reactions of 2,2,3,3,4,4,5,5-octafluoro-1,6-hexanediol were performed as per Scheme 3, and resulted in compounds 7 and 8. All the synthesized energetic polyfluoro derivatives are stable in air and can have longer shelf lives. All the compounds were confirmed by 1H and 13C NMR spectroscopy, IR spectroscopy, and elemental analysis; the obtained spectroscopic data is listed in the experimental section. The crystal structures of 4, 7 and 8 have been determined by single crystal X-ray diffraction analysis.
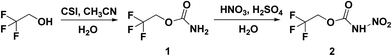 |
| | Scheme 1 Synthesis of 2,2,2-trifluoroethyl carbamate (1) and nitrocarbamate (2). | |
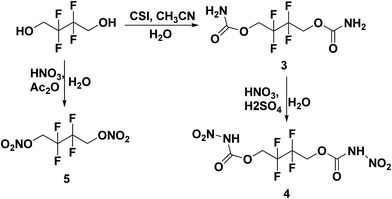 |
| | Scheme 2 Synthesis of 2,2,3,3-tetrafluorobutane-1,4-diyl dicarbamate (3), 1,4-diyl bis(nitrocarbamate) (4) and 1,4-diyl dinitrate (5). | |
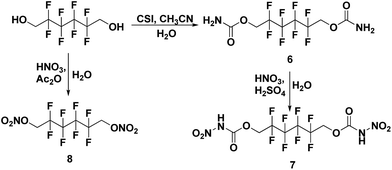 |
| | Scheme 3 Synthesis of 2,2,3,3,4,4,5,5-octafluorohexane-1,6-diyl dicarbamate (6), 1,6-diyl bis(nitrocarbamate) (7) and 1,6-diyl dinitrate (8). | |
NMR and IR spectroscopy
Compounds 2, 4, 5, 7 and 8 were investigated by multinuclear NMR spectroscopy in DMSO-d6 solvent (shown in experimental data). The 1H chemical shifts of NH and CH2 groups in compound 2 are observed at a peak at δ = 11.48 and 3.01 ppm, respectively. The 13C NMR spectrum of compound 2 shows signals at 206.9, 53.4 and 31.1 ppm for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CF3, and CH2, respectively. For compound 4, the 1H NMR spectrum shows δ = 4.78 ppm (CH2), while the 13C NMR spectrum shows peaks at 148.8, 117.9, 116.1, 115.2 and 60.1 ppm. Compound 5 displays a peak at δ = 4.66 ppm for a methylene group in the 1H NMR spectrum, and signals at 71.3 (CF2), 68.9 (CF2), and 42.4 (CH2) ppm in the 13C NMR spectrum. Compound 7 shows a peak at δ = 4.93 ppm for methylene protons and 13C NMR shifts at 148.8, 115.1, 111.0 and 60.5 ppm for CO, CF2, and CH2 carbon atoms. The 1H NMR spectrum of compound 8 shows a CH2 peak at δ = 4.66 ppm, and in the 13C NMR spectrum three signals of carbon atoms are observed at 71.3 (CF2), 68.9 (CF2) and 42.4 (CH2). The 1H and 13C NMR also indicate that molecules which have the same type of functional groups (–ONO2 and –NHNO2) show similar types of chemical shift values in the spectra. The IR spectra of compounds 2, 4 and 7 display the vibrations of the –NH2 groups in the range of 3350 and 3256 cm−1, and symmetric and asymmetric stretching vibrations in the nitro (–NHNO2) groups appear at 1611 and 1321 cm−1. The C–F stretching vibrations are observed between 1294 and 1275 cm−1. Compounds 5 and 8 show the asymmetric and symmetric stretching vibrations of the –NO2 group at 1611 and 1366 cm−1.
O, CF3, and CH2, respectively. For compound 4, the 1H NMR spectrum shows δ = 4.78 ppm (CH2), while the 13C NMR spectrum shows peaks at 148.8, 117.9, 116.1, 115.2 and 60.1 ppm. Compound 5 displays a peak at δ = 4.66 ppm for a methylene group in the 1H NMR spectrum, and signals at 71.3 (CF2), 68.9 (CF2), and 42.4 (CH2) ppm in the 13C NMR spectrum. Compound 7 shows a peak at δ = 4.93 ppm for methylene protons and 13C NMR shifts at 148.8, 115.1, 111.0 and 60.5 ppm for CO, CF2, and CH2 carbon atoms. The 1H NMR spectrum of compound 8 shows a CH2 peak at δ = 4.66 ppm, and in the 13C NMR spectrum three signals of carbon atoms are observed at 71.3 (CF2), 68.9 (CF2) and 42.4 (CH2). The 1H and 13C NMR also indicate that molecules which have the same type of functional groups (–ONO2 and –NHNO2) show similar types of chemical shift values in the spectra. The IR spectra of compounds 2, 4 and 7 display the vibrations of the –NH2 groups in the range of 3350 and 3256 cm−1, and symmetric and asymmetric stretching vibrations in the nitro (–NHNO2) groups appear at 1611 and 1321 cm−1. The C–F stretching vibrations are observed between 1294 and 1275 cm−1. Compounds 5 and 8 show the asymmetric and symmetric stretching vibrations of the –NO2 group at 1611 and 1366 cm−1.
Crystallographic data
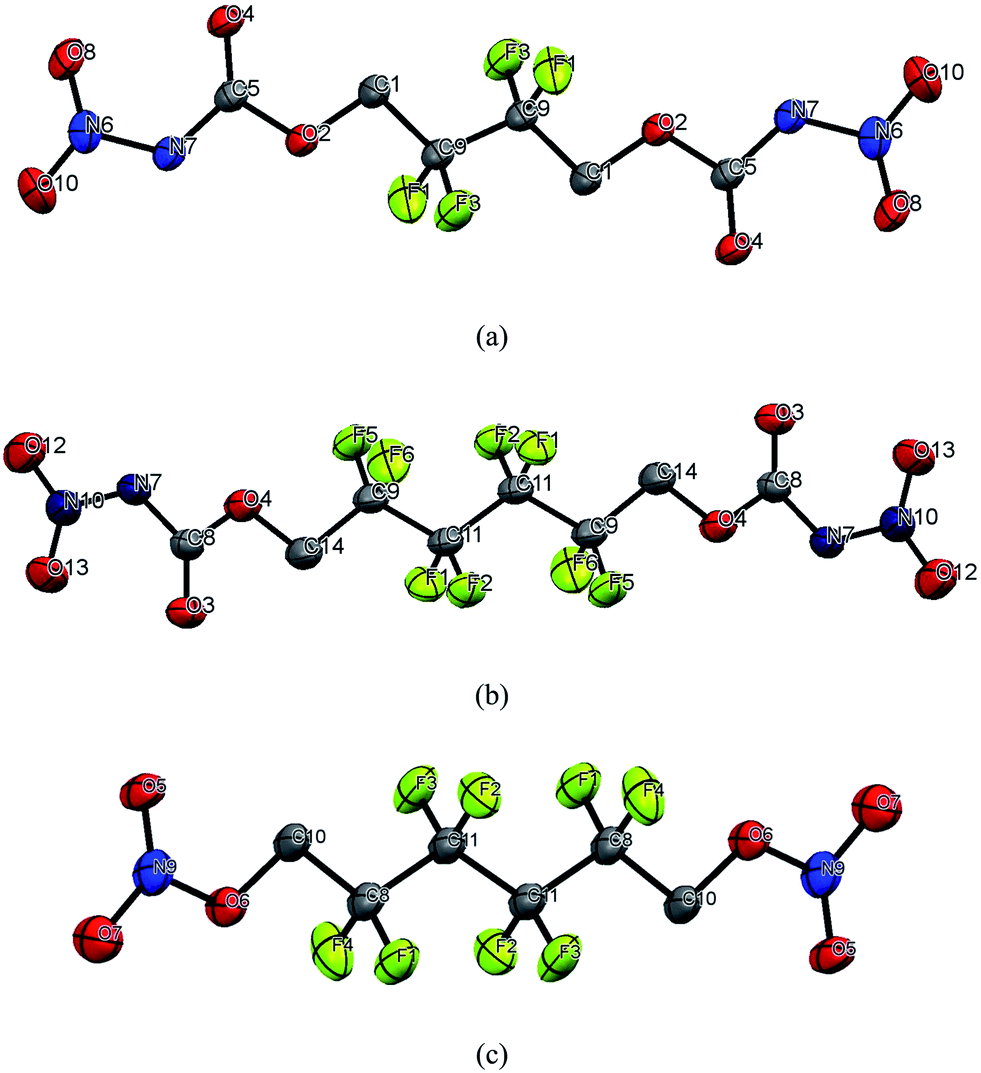
Single crystals of compounds 4 and 8 were achieved by the slow evaporation of ethyl acetate at room temperature and compound 7, from dichloromethane. All the relevant crystallographic information data is listed in Table 1. Compounds 4 and 7 crystallized in the monoclinic P21/c space group and have a density of 1.909 and 1.983 g cm−3 at 293(2) K. Compound 8 crystallized in the triclinic P1 space group with 1.924 g cm−3 density at 293(2) K. The structures of 4, 7 and 8 are shown in Fig. 1. In compound 4, the C1–O2, C5–O2, C5–N7 and N7–NO2 bond lengths are 1.434 (3) Å, 1.332 (4) Å, 1.377 (4) Å and 1.389 (4) Å, respectively. Compound 7 has C14–O4, C8–O4, C8–N7 and N7–N10 bond lengths of 1.429 (2) Å, 1.342 (3) Å, 1.391 (2) Å, and 1.377 (3) Å, respectively. The C–O and O–NO2 bond lengths in compound 8 are 1.427 (5) Å and 1.349 (3) Å, respectively. The C–F bond lengths are found between 1.343 (4) Å and 1.350 (4) Å in all the compounds.
Table 1 Crystallographic and refinement data for compounds 4, 7 and 8
| R1 = ∑||F0| − |Fc||/∑|F0|. R2 = [∑w(F02 − Fc2)2/∑w(F02)2]1/2. |
| Crystal |
4 |
7 |
8 |
| Formula |
C6H6F4N4O8 |
C8H6F8N4O8 |
C6H4F8N2O6 |
| Mr (g mol−1) |
338.15 |
438.17 |
352.11 |
| Crystal system |
Monoclinic |
Monoclinic |
Triclinic |
| Space group |
P21/c |
P21/c |
P1 |
| a (Å) |
11.4164(17) |
4.9914(16) |
5.1438(5) |
| b (Å) |
13.6481(9) |
20.7151(8) |
6.2912(9) |
| c (Å) |
4.9679(8) |
7.2618(2) |
9.9448(12) |
| α (°) |
90.00 |
90.00 |
91.60 |
| β (°) |
130.54 |
102.27 |
104.35 |
| γ (°) |
90.00 |
90 |
101.94 |
| V (Å3) |
588.28(13) |
733.72(6) |
303.96(6) |
| Z (molecules per unit cell) |
2 |
2 |
1 |
| T (K) |
293(2) |
293(2) |
293(2) |
| ρ (g cm−3) |
1.909 |
1.983 |
1.924 |
| μ (mm−1) |
0.674 |
0.663 |
0.684 |
| F(000) |
340 |
436.0 |
174.0 |
| Reflns collected |
1137 |
2526 |
1638 |
| Independent reflns |
862 |
1389 |
1126 |
| Rint |
0.0702 |
0.0848 |
0.0848 |
| Parameters |
100 |
128 |
100 |
| S on F2 |
1.138 |
1.071 |
1.071 |
| R1 [F > 2σ(F)]a |
0.0592 |
0.0467 |
0.0724 |
| wR2 (all data)b |
0.1862 |
0.1312 |
0.2273 |
| CCDC |
1437815 |
1428609 |
1424012 |
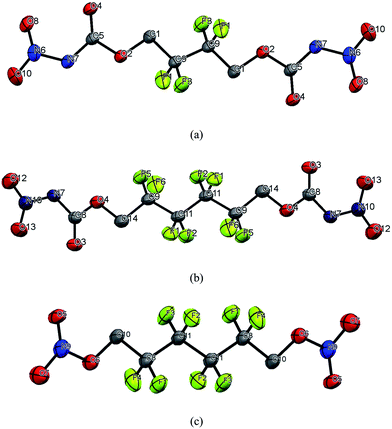 |
| | Fig. 1 Molecular units of (a) 4, (b) 7, and (c) 8. Thermal ellipsoids represent the 50% probability level. Hydrogens are not included in the structure for clarity. | |
Energetic properties
The energetic properties of compounds 2, 4, 5, 7 and 8 are listed in Table 2. The degree to which an energetic material can be oxidized is indicated by the oxygen balance (OB).12 OB is calculated using the equation proposed by Muthurajan and Ghee.13 Although all the derivatives presented in this work possess negative OB values, all of them exhibited a better oxygen balance than TNT (−74%) and TATB (−56%). As shown in Table 2, compounds 2 (−17%), 5 (−13%) and 8 (−18%) exhibit a better OB than that of RDX (−21.6%) and HMX (−21.6%). Meanwhile, compounds 4 (−23%) and 7 (−25%) show a slightly lower OB than RDX. The densities of compounds 4, 7 and 8 were obtained from X-ray crystallography analysis while compounds 2 and 5 were measured by a gas pycnometer. The densities of these compounds were found to be between 1.88–1.98 g cm−3 and higher than those of TNT (1.65 g cm−3) and RDX (1.80 g cm−3). The high values of densities place these compounds in a class of relatively denser compounds. Among these compounds, compound 7 has the highest density of 1.98 g cm−3. The high densities in these molecules are due to fluorine and the extensive intra- and inter-molecular hydrogen bonds. The heat of formation (HOF) is the fundamental property in determining the performance of explosives. All the compounds show negative HOFs due to the presence of fluorine in the structure. EXPLO5 v6.01 (ref. 14) was used to calculate the detonation properties. The detonation velocities (D) of compounds 2–8 vary from 6.68 to 8.05 km s−1, whereas the detonation pressures (P) fall in the range of 20.55 to 36.79 GPa. Compounds 2–5 show a superior performance relative to TNT (D = 6.88 km s−1 and P = 19.5 GPa). The thermal stabilities of these compounds were determined by thermogravimetric-differential thermal analysis (TG-DTA) measurements (Table 2) at a heating rate of 5 °C min−1. Compounds 4, 5 and 8 decompose without melting at 188, 151 and 202 °C, respectively. Compound 7 melts at 124 °C and starts decomposing at 173 °C. Overall, considering the performance parameters and thermal stabilities, compounds 7 and 8 are promising energetic materials. Due to the energetic nature of these compounds, impact and friction sensitivities were measured and listed in Table 2. All the compounds except 5 (IS = 35 J) show impact sensitivity > 40 J, while friction sensitivity (FS) > 360 N, and are classified as insensitive explosives.
Table 2 Energetic properties of fluorine derivatives relative to TNT and RDX
| Compd |
OBa (%) |
HOFb (kJ mol−1) |
Density (g cm−3) |
Dc (km s−1) |
Pd (GPa) |
Tme (°C) |
Tdecf (°C) |
ISg (J) |
FSh [N] |
| Calculated oxygen balance. Calculated heat of formation. Predicted detonation velocity. Predicted detonation pressure. Melting point. Decomposition temperature. Impact sensitivity. Friction sensitivity. Density measured by gas pycnometer. Experimental crystal density. Ref. 16. Ref. 12. Ref. 17. |
| 2 |
−17.0 |
−1114.4 |
1.88i |
7.37 |
20.55 |
Dec |
192.1 |
>40 |
>360 |
| 4 |
−23.7 |
−1639.5 |
1.90j |
7.22 |
24.00 |
Dec |
188.78 |
>40 |
>360 |
| 5 |
−12.7 |
−1218.9 |
1.94i |
8.05 |
36.79 |
Dec |
151.14 |
35 |
>360 |
| 7 |
−25.6 |
−2535.9 |
1.98j |
6.68 |
25.26 |
124.93 |
173.07 |
>40 |
>360 |
| 8 |
−18.2 |
−2082.2 |
1.92j |
6.71 |
27.36 |
Dec |
202.21 |
>40 |
>360 |
| TNT |
−74.0 |
−67.0k |
1.65k |
6.88k |
19.5k |
80.8l |
295k |
15k |
>353l |
| RDX |
−21.6 |
63.0l |
1.80l |
8.60l |
33.92l |
205l,m |
230k |
7.5m |
120m |
Conclusions
The syntheses of 2,2,2-trifluoroethyl nitrocarbamate (2), 2,2,3,3-tetrafluorobutane-1,4-diyl bis(nitrocarbamate) (4), 2,2,3,3-tetrafluorobutane-1,4-diyl dinitrate (5), 2,2,3,3,4,4,5,5-octafluorohexane-1,6-diyl bis(nitrocarbamate) (7), and 2,2,3,3,4,4,5,5-octafluorohexane-1,6-diyl dinitrate (8) were carried out and their energetic properties were determined. These compounds displayed high densities (1.88–1.98 g cm−3) with decomposition temperatures above 150 °C. All the compounds exhibited detonation velocities above 6.68 km s−1 and pressures over 20.55 GPa. Moreover, all the compounds were observed to be insensitive toward impact (35–40 J) and friction (>360 N). Considering their high densities, reasonable detonation performances, insensitive nature, and thermal stabilities, these compounds have potential as high energy density materials.
Experimental section
Safety precautions
Although, we have experienced no difficulties during the syntheses and characterization of these materials, proper protective measures should be used. Appropriate safety precautions, such as the use of shields in a fume hood and personal protection equipment (safety glasses, face shields, ear plugs, gloves and leather and/or Kevlar suits) should be taken all the time when handling these materials.15 Scratching, scraping or similar mechanical actions must be avoided. Ignoring safety precautions may lead to serious injuries.
General methods
1H and 13C NMR spectroscopic data were recorded at 400 MHz and 101 MHz, respectively, with the solvent resonance as an internal standard (for DMSO-d6, 1H NMR at δ = 2.50 and 3.50 ppm; 13C NMR at δ = 44.0 ppm). Chemical shifts were reported relative to tetramethylsilane at 0.00 ppm. Elemental analyses (C, H, and N) were performed on a flash EA 1112 full automatic trace element analyser. Densities were measured at room temperature using a Micromeritics Accupyc 1330 gas pycnometer. Melting and decomposition points were recorded on a differential scanning calorimeter (DSC, TA Instruments Q10) at a scan rate of 5 °C min−1. Impact and friction sensitivities were measured by employing a standard BAM Fallhammer apparatus and a BAM friction tester. The IR spectra were recorded on a Perkin-Elmer IR spectrometer using KBr pellets.
2,2,2-Trifluoroethyl carbamate (1). To a dry acetonitrile (20 mL) solvent placed in an ice bath at 0 °C, 2,2,2-trifluoroethanol (1 g, 7.54 mmol) was suspended, and then, chlorosulfonyl isocyanate CSI (1.38 g, 6.02 mmol) was added drop by drop with caution. 2,2,2-Trifluoroethanol was dissolved during the addition and after complete addition a colourless precipitate appeared. After that, the ice bath was removed and the mixture was stirred for 1 h at ambient temperature. The reaction mixture was again kept in an ice bath and 10 mL of water added slowly. After phase separation, the organic solvent was removed under reduced pressure and the resulting precipitate was filtered. The precipitate was washed with cold water and dried under reduced pressure yielding pure 2,2,2-trifluoroethyl carbamate (1). White solid (0.800 g, 74% yield). 1H NMR: δ = 7.12 (s, 1H), 2.08 (s, 2H); 13C NMR: δ = 206.9, 53.4, 31.1; IR (KBr pellet): 1717, 1401, 1216, 1062, 1007, 773, 618, 578 cm−1; elemental analysis (%) calcd for C3H4F3NO2 (143.02): C, 25.19; H, 2.82; N, 9.79. Found: C, 25.40; H, 2.50; N, 9.50.
2,2,2-Trifluoroethyl nitrocarbamate (2). To a mixture of fuming HNO3 (99.5%, 4 mL) and concentrated H2SO4 (4 mL) at 0 °C, 2,2,2-trifluoroethyl carbamate (1) (0.400 g, 2.79 mmol) was added slowly. The mixture was stirred at 0 °C for 10 min and then at ambient temperature for 1 h. The reaction mixture was poured onto 100 g of ice. The resulting precipitate was filtered, washed with water and dried under vacuum to get colorless 2,2,2-trifluoroethyl nitrocarbamate (2). White solid (0.300 g, 57% yield). 1H NMR: δ = 11.48 (s, NH), 3.01 (s, 2H); 13C NMR: δ = 206.9, 53.4, 31.1; IR (KBr pellet): 3256, 1747, 1611, 1420, 1321, 1275, 1209, 1185, 1147, 1121, 953, 942, 923, 841, 752 cm−1; elemental analysis (%) calcd for C3H3F3N2O4 (188.00): C, 19.16; H, 1.61; N, 14.90; found: C, 19.25; H, 1.50; N, 14.50.
2,2,3,3-Tetrafluorobutane-1,4-diyl dicarbamate (3). Compound 3 was prepared by the reaction of 2,2,3,3-tetrafluorobutane-1,4-diol (0.550 g, 3.39 mmol) and chlorosulfonyl isocyanate CSI (0.73 g, 8.48 mmol), using a procedure identical to that used for the synthesis of 1. White solid (0.750 g, 89% yield). 1H NMR: δ = 7.07 (s, NH2), 6.84 (s, NH2), 4.54 (t, 4H); 13C NMR: δ = 155.5, 117.9, 116.1, 115.9, 59.2; IR (KBr pellet): 3425, 1717, 1607, 1415, 1364, 1279, 1188, 1141, 1104, 1059, 999, 954, 910, 779, 748 cm−1; elemental analysis (%) calcd for C6H8F4N2O4 (248.04): C, 29.04; H, 3.25; 11.29; found: C, 29.30; H, 3.40; N, 11.52.
2,2,3,3-Tetrafluorobutane-1,4-diyl bis(nitrocarbamate) (4). Compound 4 was prepared using fuming HNO3 (99.5%, 4 mL), concentrated H2SO4 (4 mL) and 2,2,3,3-tetrafluorobutane-1,4-diyl dicarbamate 3 (0.420 g, 1.0 mmol) by a similar procedure as used for the synthesis of compound 2. White solid (0.271 g, 48% yield). 1H NMR: δ = 11.19 (s, NH), 4.78 (t, 2H); 13C NMR: δ = 148.8, 117.9, 116.1, 115.2, 60.1; IR (KBr pellet): 3276, 1767, 1611, 1446, 1323, 1289, 1209, 1174, 1147, 1101, 993, 962, 923, 841, 752, 696 cm−1; elemental analysis (%) calcd for C6H6F4N4O8 (338.01): C, 21.31; H, 1.79; N, 16.57; found: C, 21.52; H, 1.92; N, 16.65.
2,2,3,3-Tetrafluorobutane-1,4-diyl dinitrate (5). To a mixture of acetic acid (12 mL) and acetic anhydride (12 mL), HNO3 (8.0 g, 98%) was added carefully at 0 °C. The reaction mixture was stirred for 20 min and 2,2,3,3-tetrafluorobutane-1,4-diol (0.550 g, 3.39 mmol) was added in small portions. The stirring of the reaction mixture was continued for 3 h at 20 °C and then poured onto ice/water (200 mL). The resulting precipitate was filtered, washed with water and dried under vacuum to yield colourless 2,2,3,3-tetrafluorobutane-1,4-diyl dinitrate (5). White solid (0.450 g, 53% yield). 1H NMR: δ = 4.66 (s, 2H); 13C NMR: δ = 71.3, 68.9, 42.4; IR (KBr pellet): 2229, 1715, 1611, 1420, 1366, 1280, 1193, 956, 779, 711, 676, 595 cm−1; elemental analysis (%) calcd for C4H4F4N2O6 (252): C, 19.06; H, 1.60; N, 11.11; found: C, 19.20; H, 1.82; N, 11.20.
2,2,3,3,4,4,5,5-Octafluorohexane-1,6-diyl dicarbamate (6). Compound 6 was prepared by the reaction of 2,2,3,3,4,4,5,5-octafluorohexane-1,6-diol (0.500 g, 1.90 mmol) with chlorosulfonyl isocyanate CSI (0.415 g, 2.93 mmol) by a similar procedure as used for the synthesis of compound 1. White solid (0.400 g, 55% yield). 1H NMR: δ = 7.12 (s, NH2), 6.92 (s, NH2), 4.67 (t, 2H); 13C NMR: δ = 155.2, 118.1, 115.6, 111.2, 59.0; IR (KBr pellet): 3441, 3218, 2973, 1705, 1613, 1419, 1366, 1281, 1118, 955, 836, 779, 711, 676 cm−1; elemental analysis (%) calcd for C8H8F8N2O4 (348.04): C, 27.60; H, 2.32; N, 8.05; found: C, 27.82; H, 2.40; N, 8.60.
2,2,3,3,4,4,5,5-Octafluorohexane-1,6-diyl bis(nitrocarbamate) (7). Compound 7 was prepared by the reaction of 2,2,3,3,4,4,5,5-octafluorohexane-1,6-diyl dicarbamate 6 (0.500 g, 1.3 mmol) with fuming HNO3 (99.5%, 4 mL) and concentrated H2SO4 (4 mL) by a similar procedure as used for the synthesis of compound 2. White solid (0.400 g, 65% yield). 1H NMR: δ = 4.93 (t, 2H); 13C NMR: δ = 148.8, 115.1, 111.0, 60.5; IR (KBr pellet): 3266, 1782, 1612, 1444, 1324, 1294, 1178, 1121, 1019, 984, 957, 850, 807, 749, 697 cm−1; elemental analysis (%) calcd for C8H6F8N4O8 (438.14): C, 21.93; H, 1.38; N, 12.79; found: C, 21.81; H, 1.40; N, 12.82.
2,2,3,3,4,4,5,5-Octafluorohexane-1,6-diyl dinitrate (8). Compound 8 was prepared using HNO3 (8.0 g, 98%), acetic acid (12 mL), acetic anhydride (12 mL) and 2,2,3,3,4,4,5,5-octafluorohexane-1,6-diol (0.500 g, 1.90 mmol) by a similar procedure as used for the synthesis of compound 5. White solid (0.350 g, 57% yield). 1H NMR: δ = 4.66 (s, 2H); 13C NMR: δ = 71.3, 68.9, 42.4; IR (KBr pellet): 2229, 1715, 1611, 1420, 1366, 1280, 1193, 956, 779, 711, 676, 595 cm−1; elemental analysis (%) calcd for C6H4F8N2O6 (351.99): C, 20.47; H, 1.15; N, 7.96; found: C, 20.60; H, 1.20; N, 8.00.
Acknowledgements
DS thanks Dr K. Muralidharan, School of Chemistry, University of Hyderabad, India for providing experimental facilities. DS is also grateful to Prof. J. M. Shreeve, Department of Chemistry, University of Idaho for providing experimental and computational facilities. VDG thanks the DST-SERB for the research grant (Young Scientists, No. SB/FT/CS-110/2014).
References
- Q. Zhang and J. M. Shreeve, Chem. Rev., 2014, 114, 10527–10574 CrossRef CAS PubMed; H. Gao and J. M. Shreeve, Chem. Rev., 2011, 111, 7377–7436 CrossRef PubMed.
- H. Gao, C. Ye, R. W. Winter, G. L. Gard, M. E. Sitzmann and J. M. Shreeve, Eur. J. Inorg. Chem., 2006, 3221–3226 CrossRef CAS; R. P. Singh, R. W. Winter, G. L. Gard, Y. Gao and J. M. Shreeve, Inorg. Chem., 2003, 42, 6142–6146 CrossRef PubMed; S. Garg and J. M. Shreeve, J. Mater. Chem., 2011, 21, 4787–4795 RSC.
- R. P. Singh, S. Manandhar and J. M. Shreeve, Tetrahedron Lett., 2002, 43, 9497–9499 CrossRef CAS; T. Abe, Y. H. Joo, G. H. Tao, B. Twamley and J. M. Shreeve, Chem.–Eur. J., 2009, 15, 4102–4110 CrossRef PubMed; T. Abe, G. H. Tao, Y. H. Joo, Y. Huang, B. Twamley and J. M. Shreeve, Angew. Chem., Int. Ed., 2008, 47, 7087–7090 CrossRef PubMed.
- Y. R. Mirzaei, B. Twamley and J. M. Shreeve, J. Org. Chem., 2002, 67, 9340–9345 CrossRef CAS PubMed; L. Xu, W. Chen, J. F. Bickley, A. Steiner and J. Xiao, J. Organomet. Chem., 2000, 596, 409–416 CrossRef; Z. Zeng, B. S. Phillips, J. C. Xiao and J. M. Shreeve, Chem. Mater., 2008, 20, 2719–2726 CrossRef; A. S. Kumar, N. Kommu, V. D. Ghule and A. K. Sahoo, J. Mater. Chem. A, 2014, 2, 7917–7926 Search PubMed.
- R. D. Verma, R. L. Kirchmeier and J. M. Shreeve, Adv. Inorg. Chem., 1994, 41, 125–169 CrossRef CAS; Y. Huang, G. L. Gard and J. M. Shreeve, Tetrahedron Lett., 2010, 51, 6951–6954 CrossRef; H. Xue and J. M. Shreeve, Eur. J. Inorg. Chem., 2005, 2573–2780 CrossRef; D. Srinivas, V. D. Ghule, S. P. Tewari and K. Muralidharan, Chem.–Eur. J., 2012, 18, 15031–15037 CrossRef PubMed.
- Y. Suzuki, T. Hagiwara, I. Kawamura, N. Okamura, T. Kitazume, M. Kakimoto, Y. Imai, Y. Ouchi, H. Takezoe and A. Fukuda, Liq. Cryst., 2006, 33, 1344–1349 Search PubMed; J. A. Smith Jr, R. A. Distasio, N. A. Hannah, R. W. Winter, T. J. R. Weakley, G. L. Gard and S. B. Rananavare, J. Phys. Chem. B, 2004, 108, 19940–19948 CrossRef CAS; P. Kirsch, M. Bremer, A. Taugerbeck and T. Wallmichrath, Angew. Chem., Int. Ed., 2001, 40, 1480–1484 CrossRef.
- Y. R. Mirzaei, B. Twamley and J. M. Shreeve, J. Org. Chem., 2002, 67, 9340–9345 CrossRef CAS PubMed; H. Xue and J. M. Shreeve, Eur. J. Inorg. Chem., 2005, 2573–2580 CrossRef.
- C. Ye, G. L. Gard, R. W. Winter, R. G. Syvret, B. Twamley and J. M. Shreeve, Org. Lett., 2007, 9, 3841–3844 CrossRef CAS PubMed.
- R. Haiges, C. B. Jones and K. O. Christe, Inorg. Chem., 2013, 52, 5551–5558 CrossRef CAS PubMed; R. Haiges and K. O. Christe, Dalton Trans., 2015, 44, 10166 RSC; K. O. Christe, W. W. Wilson, G. Blanger-Chabot, R. Haiges, J. A. Boatz, M. Rahm, G. K. Surya Prakash, T. Saal and M. Hopfinger, Angew. Chem., Int. Ed., 2015, 54, 1316–1320 CrossRef PubMed.
- M. A. Kettner and T. M. Klapötke, Chem.–Eur. J., 2015, 21, 3755–3765 CrossRef CAS PubMed.
- J. K. Rasmussen and A. Hassner, Chem. Rev., 1976, 76, 389–408 CrossRef CAS; Q. J. Axthammer, B. Krumm and T. M. Klapötke, Eur. J. Org. Chem., 2015, 723–729 CrossRef.
- R. Meyer, J. Köhler and A. Homburg, Explosives, Wiley-VCH, 5th edn, 2002 Search PubMed.
- H. Muthurajan and A. H. Ghee, Cent. Eur. J. Energ. Mater., 2008, 5, 19–35 CAS.
- M. Sućeska, Propellants, Explos., Pyrotech., 1991, 16, 187–197 CrossRef.
- R. Haiges, J. A. Boatz, M. Yousufuddin and K. O. Christe, Angew. Chem., Int. Ed., 2007, 46, 2869–2874 CrossRef CAS PubMed.
- V. Thottempudi and J. M. Shreeve, J. Am. Chem. Soc., 2011, 133, 19982–19992 CrossRef CAS PubMed.
- D. M. Badgujar, M. B. Talawar, S. N. Asthana and P. P. Mahulikar, J. Hazard. Mater., 2008, 151, 289–305 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1424012, 1428609 and 1437815. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra24615f |
| ‡ Current address: Department of Chemistry, University of Idaho, Moscow, Idaho 83844-2343, USA, E-mail: E-mail: dharavathsrinivas@gmail.com |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.