
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel non-hydrolytic templated sol–gel synthesis of mesoporous aluminosilicates and their use as aminolysis catalysts†
David
Skoda
ab,
Ales
Styskalik
ab,
Zdenek
Moravec
a,
Petr
Bezdicka
c,
Michal
Babiak
ab,
Mariana
Klementova
c,
Craig E.
Barnes
d and
Jiri
Pinkas
 *ab
*ab
aDepartment of Chemistry, Masaryk University, Kotlarska 2, CZ-61137 Brno, Czech Republic. E-mail: jpinkas@chemi.muni.cz
bCEITEC MU, Masaryk University, Kamenice 5, CZ-62500 Brno, Czech Republic
cInstitute of Inorganic Chemistry of the ASCR, v.v.i., CZ-25068 Husinec-Rez, Czech Republic
dDepartment of Chemistry, University of Tennessee, Knoxville, TN 37996-1600, USA
First published on 25th February 2016
Abstract
A novel non-hydrolytic sol–gel (NHSG) synthesis of mesoporous aluminosilicate xerogels is presented. The polycondensation between silicon acetate, Si(OAc)4, and tris(dimethylamido)alane, Al(NMe2)3, leads to homogeneous aluminosilicate xerogels containing Si–O–Al linkages through dimethylacetamide elimination. The addition of Pluronic P123 and F127 templates provides stiff gels that are, after calcination at 500 °C, converted to stable mesoporous xerogels with a high surface area (>600 m2 g−1) and wormhole-type pores (d = 5.9 nm). The xerogels exhibit high catalytic activity in aminolysis of styrene oxide (82% conversion) with the turnover frequency up to 100.
Introduction
Aluminosilicate zeolites with their uniform molecular-sized pores, high surface acidity, and large number of active sites have been extensively applied as molecular sieves, ion exchangers, and solid acid catalysts in industrial catalytic processes since the last century.1,2 They are usually prepared from alkoxide precursors under hydrothermal conditions by so-called molecular templating wherein a cationic organic species acts as a template for the formation of ordered pores and channels. This approach generates highly ordered microporous materials whose pore diameters are controlled by the size of the molecular templates. The pore sizes of zeolites are therefore below 15 Å and typically 4–10 Å.3 However, the conversion or adsorption of bulky organic molecules requires large pore channels (>2 nm), which has not been achieved in stable microporous zeolites. A synthetic challenge in this context is the disparate hydrolysis rates of Si and Al alkoxides which lead to precipitation of alumina rich phases and has to be prevented by prehydrolysis4 or chelation methods.5 The discovery of ordered mesoporous silicates, such as MCM-41, with high surface areas, well-defined and tunable pore diameters from 1.5 to 10 nm was a significant advancement in this field. The catalysts with the hexagonal structure, such as Al–MCM-41, offered an opportunity for processing bulky molecules6 and attracted much interest.7 Unfortunately, the poor hydrothermal stability of the MCM-41-type materials restricts their wider applications in industrial catalysis. Nevertheless, improved hydrothermal stability of cubic Al–MCM-48 materials was achieved by supercritical fluid (SCF) impregnation.8 Increase of hydrothermal stability was achieved also by 2-D hexagonal mesoporous SBA-15 materials9 with large pores (4.6–30 nm), thick pore walls (3.1–6.4 nm), and high specific surface areas (up to 1000 m2 g−1). However, electrically neutral frameworks of silica SBA-15 materials exhibit very weak acidity derived from the silanol groups located on the pore walls. Therefore, much effort have been devoted to increasing the acidity by introduction of Al into the framework by direct synthesis,10 post-synthesis11 and grafting or impregnation12 strategies. The procedure of direct synthesis is relatively simple, but only a small number of the heteroatoms can be introduced into the mesophases. By contrast, the procedure of grafting or impregnation is complicated and difficult to replicate. Moreover, there are always more 6-coordinated Al atoms in these materials than 4-coordinated ones which are essential for Brønsted acidity.13 Al–SBA-15 materials with a large number of moderate acidic sites are very promising catalysts and supports allowing catalytic cracking, alkylation, and hydrotreating reactions.14,15 The preparation of Al–SBA-15 via the post-synthesis method was reported by Li16 by treatment with aluminum isopropoxide. Han reported well-ordered hexagonal mesoporous aluminosilicates synthesized by the re-crystallization of SBA-15 in a dilute solution of aluminosilicate sol, glycerol, or both.17 Recently, the assembly of zeolite seeds or coating of walls of mesoporous matrices (SBA-15, MCM-48) with zeolites was described as a promising way to novel class of hydrothermally stable mesoporous aluminosilicate catalysts. The assembly of ZSM-5, beta, TS-1, and FAU zeolite seeds was achieved with the use of structure-directing agents, such as cetyltrimethyl ammonium bromide (CTAB) or Pluronic P123.18–21 A so-called coated route approach using a dilute clear solution of primary zeolite units was employed to synthesize ultrastable mesoporous aluminosilicate molecular sieves.22,23 This method produces a new type of nanocrystalline zeolitic material with mesopore walls formed by a templated solid-state secondary crystallization of zeolites into walls of SBA-15.Recently, Liu13 has obtained Al-rich mesoporous aluminosilicates with improved hydrothermal stability by assembling zeolite Y and beta precursors in the walls of mesophases. For example, the retaining ratio of the total surface area was 33% after hydrothermal treatment in 100% water vapor at 800 °C for 15 h. This group also reported a pH-adjusting method24 based on the assembly of beta zeolite. An approach recently published by Enterría25 provides a simple strategy based on the overgrowing mesoporous silica (MCM-48) on crystalline zeolite particles. For this purpose, preformed zeolite is added to mesoporous silica gel, which results in materials with a microporous zeolitic core and a mesoporous silica cover (MCM-48).
The methods and procedures mentioned above all involve hydrolytic reactions. Aluminosilicates can also be successfully prepared by non-hydrolytic routes. With these methods, it is possible to overcome the problems of different hydrolysis rates and subsequent phase separation. These techniques allow reaction control on atomic scale and homogeneous dispersions of silicon and aluminum atoms in the gel network may be achieved. One type of non-hydrolytic sol–gel (NHSG) reaction is alkylhalogenide elimination.26 In this method, condensations between SiCl4, AlCl3, Al(OiPr)3, and Si(OiPr)4 in ethers or CH2Cl2 provide monolithic homogeneous aluminosilicate gels27 (Scheme 1).
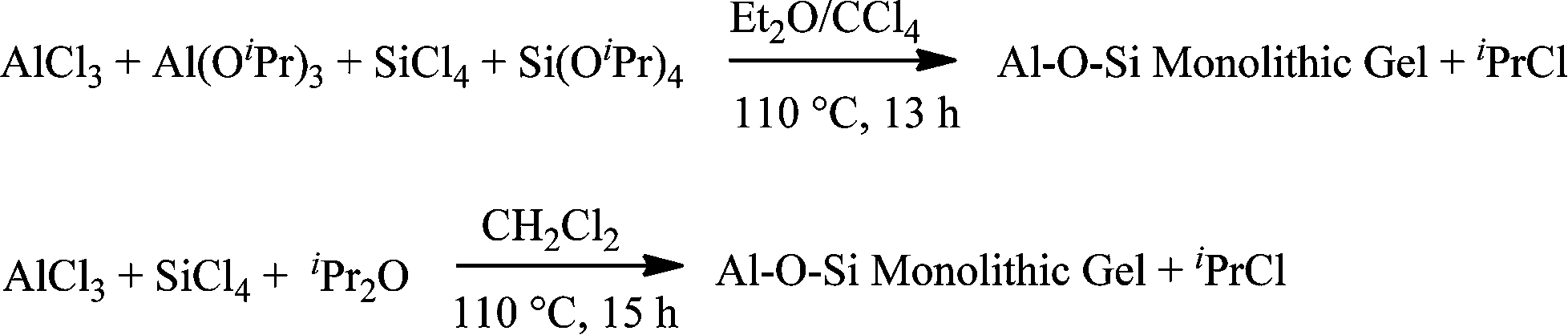 | ||
| Scheme 1 Non-hydrolytic alkylhalogenide elimination. | ||
This non-hydrolytic sol–gel technique has also been applied to the preparation of highly active mesoporous metathesis catalysts MoO3–SiO2–Al2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 28 and Re2O7–SiO2–Al2O3.29 Mullite precursor gel synthesis was described from AlCl3 and TEOS in Et2O and CCl4 in an autoclave at 110 °C.30
28 and Re2O7–SiO2–Al2O3.29 Mullite precursor gel synthesis was described from AlCl3 and TEOS in Et2O and CCl4 in an autoclave at 110 °C.30
Catalytic efficiency of aluminosilicates was determined by a variety of test reactions. Kim reported Friedel–Crafts acylation of bulky aromatic compounds in MFI zeolite nanosponge,31 Neves32 used mesoporous aluminosilicates with acid sites for conversion of furfuryl alcohol to ethyl levulinate and Robinson described the alcoholysis and aminolysis of styrene oxide promoted by mesoporous aluminosilicate catalysts.33,34
We have recently reported that non-hydrolytic acetamide elimination35 can be successfully used for the synthesis of mesoporous titanium silicate36 and zirconium silicate materials.37 In contrast to conventional hydrolysis-based routes for the preparation of SBA-15 and MCM-41 materials, our approach excludes hydrolytic steps and the reactions proceed only by condensation of metal amide with silicon acetate.
The work presented here introduces this novel non-hydrolytic acetamide elimination for the preparation of aluminosilicate xerogels with a high Al content in the framework. This effective one-pot reaction can be extended by the use of templates. With the Pluronic block copolymers we are able to obtain homogeneous xerogels which exhibit their mesoporous character even after calcination at 700 °C and convert to mullite only at 980–1000 °C. We have successfully used these materials as catalysts for aminolysis34,38 and alcoholysis33 of styrene oxide and for the conversion of styrene oxide to phenylacetaldehyde.39
Experimental
General procedures
All manipulations were performed in dry nitrogen using Schlenk techniques or in an M. Braun drybox with both H2O and O2 levels below 1 ppm. Al(NMe2)3 is highly flammable and reacts violently with water, liberating flammable gases. It must be handled under dry inert gas at all times.Chemicals, such as Pluronic P123 (EO20PO70EO20Mav = 5845 g mol−1), Pluronic F127 (EO100PO65EO100Mav = 12600 g mol−1), tris(dimethylamido)aluminium Al(NMe2)3 (99%), styrene oxide (97%), aniline (97%), methanol, and nonane, were purchased from Sigma-Aldrich. Si(OAc)4, Si2O(OAc)6, Me3SiOAc, and Si2(CH2)2(OAc)6 were synthesized according to the published procedures.40 Toluene, methanol, and isopropanol were dried by standard methods and distilled before use. Pluronic P123 and F127 were dried under vacuum at 60 °C, dissolved in dry toluene, and stored as 20.17 and 19.61 wt% solutions, respectively.
Characterization
IR spectra were recorded on a Bruker Tensor 27 FTIR spectrometer from KBr pellets or on a Bruker Alpha-Platinum ATR system. GC-MS measurements were performed on a mass spectrometer TSQ Quantum XLS coupled with a gas chromatograph Trace GC Ultra by Thermo Scientific. The gas chromatograph was equipped with a TS-SQC column (length 15 m, diameter 0.25 mm, film thickness 0.25 μm) using a temperature program: 50 °C (0 min), 5 °C min−1 to 80 °C, 15 °C min−1 ramp to 120 °C, 35 °C min−1 ramp to 200 °C, held at 200 °C for 0.5 min. Split injection mode was used with the injector, interface, and detector temperatures all set to 200 °C. The column pressure was 31.5 kPa and the ionization energy was 70 eV. DIP MS measurements were performed with the temperature program 30 °C for 30 s, 50 °C min−1 30–450 °C, and held at this temperature for 1 min; the ionization energy was 44 eV. For liquid phase 1H and 13C NMR measurements, a Bruker Avance II DRX 300 MHz spectrometer was used. The spectra were referenced to the residual proton or carbon resonances of benzene-d6 (7.15 and 128.0 ppm) and CDCl3 (7.20 and 77.0 ppm). Solid state NMR spectra were measured on a Bruker Avance III 700 MHz spectrometer with a MAS DVT 700S4 BL4 N-P/H probe and on a Bruker Avance III 500 MHz spectrometer with a MAS VTN 500SB BL4 N-P/F-H probe. Chemical shifts were referenced externally to 29Si δ [(Me3SiO)8Si8O20]: 11.72 ppm; 13C δ [adamantane]: 38.68 ppm; 27Al δ [[Al(H2O)6]3+ (aq. solution)]: 0.0 ppm. High temperature PXRD diffractograms were recorded on an X'PertPRO diffractometer equipped with a CoKα X-ray tube and a HTK 16 high-temperature chamber (Anton Paar, Graz, Austria) with a Pt holder. Samples were measured from 500 to 1250 °C in 50 °C increments. The sample was held during scanning at a constant temperature for 12 min. Nitrogen adsorption/desorption experiments were performed at 77 K on a Quantachrome Autosorb-1MP porosimeter. Prior to the measurements, the samples were degassed at 100 °C for at least 24 h until the outgas rate was less than 0.4 Pa min−1. The adsorption–desorption isotherms were measured for each sample at least three times. The specific surface areas (SA), total pore volumes (Vtot at p/p0 = 0.98) and average pore size (d) were determined by volumetric techniques41,42 and analyzed by the multipoint BET method using at least five data points. The valid pressure ranges were established by the Rouquerol transform and equivalent BET surface areas were calculated. Pore size distributions were calculated by the NLDFT method from the adsorption branch of the isotherms with the use of a nitrogen-on-silica kernel. Thermal analysis (TG/DSC) was performed on a Netzsch STA 449C Jupiter apparatus in the stream of air (70 cm3 min−1) with a temperature ramp of 5 °C min−1 to 1000 °C, in a Pt crucible. Aluminium and silicon contents were determined on an ICP optical emission spectrometer iCAP 6500 Duo (Thermo, UK) equipped with a solid-state generator with a frequency of 27.12 MHz and a maximum power input 1350 W. The measurements of Al were performed at 167.0, 308.2, and 309.2 nm. For Si analysis, wavelengths 212.4 and 251.6 nm were used. Transmission electron microscopy (TEM) images were obtained from a JEOL JEM 3010 microscope operated at 300 kV (LaB6 cathode, point resolution 1.7 Å) with an Oxford Instruments Energy Dispersive X-ray (EDX) detector attached. SAXS measurements were performed on a Rigaku BioSAXS 1000 system at wavelength of 1.5408 Å with a PILATUS 100K detector, Dectris Lts. Scattering curves were analyzed by ATSAS software package. Studies of surface acidity were performed on air-calcined xerogels. Xerogels (ca. 50 mg) were dried before adsorption under vacuum (1 h, 115 °C). Then, the samples were exposed for 30 min to pyridine vapors under its autogenous pressure at 25 °C. After adsorption, samples were dried at room temperature under vacuum for 2 h. Adsorbed pyridine was characterized by the IR technique.The single-crystal X-ray diffraction data were collected at 120 K using Rigaku diffraction system (MicroMax007HF DW rotating anode source with multilayer optic, partial χ axis goniometer, Saturn 724+ HG detector and Cryostream cooling device). Molybdenum Kα radiation (λ = 0.71075 Å) was used. CrystalClear (Rigaku 2014) and CrysAlisPro (Agilent Technologies 2013) software packages were used for data collection and data reduction. The structure was solved using SHELXT43 program and refined (full matrix least-squares refinement on F2) using SHELXL program. Both symmetrically independent cation fragments were disordered. One Me3Si-moiety of one anionic fragment was also refined as disordered. Disordered fragments were treated by geometrical similarity restraints and by ADP restraints. All non-hydrogen atoms were refined anisotropically. All hydrogen atoms (of methyl and hydroxyl groups) were placed at calculated positions and refined as riding and rotating, with their Uiso set to 1.5Ueq of carrier atom.
In reactions without a template, the yield of the product as well as the mass of starting precursors were precisely weighed to allow gravimetric estimation of the degree of condensation, DC = 100(ntotal − nresidual)/ntotal, where ntotal is the molar amount of organic groups in the starting materials and nresidual is molar amount of residual organic groups in the xerogel based on the difference between theoretical and experimental yields. As the condensation reactions were never quantitative, the degree of condensation represents the relative difference between the maximum theoretical loss of Me2NC(O)CH3 (eqn (1)) in comparison to what is experimentally observed. This difference also defines the number of acetoxy groups on silicon and dimethylamido groups on aluminium that are left in the matrix.
Aluminosilicate xerogel prepared without template SiAl1 (eqn (1))
Al(NMe2)3 (0.799 g; 5.02 mmol) dissolved in 15 cm3 of dry toluene was added dropwise by a syringe to a stirred solution of Si(OAc)4 (1.033 g; 3.907 mmol) in 15 cm3 of dry toluene at 80 °C. After the addition, a yellow gel formed. The reaction mixture was heated to 80 °C for 7 days, after which the volatile byproducts were separated under vacuum and the yellow powder was dried under vacuum for 24 h. Yield 1.098 g, theor. 0.494 g; DC = 56%.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1596 vs (ν COO), 1536 vs (ν COO), 1718 vw (ν COO), 2799 vw (ν CH3), 2930 w (ν CH3), 3410 vw (ν NH).
O), 1596 vs (ν COO), 1536 vs (ν COO), 1718 vw (ν COO), 2799 vw (ν CH3), 2930 w (ν CH3), 3410 vw (ν NH).
NMR: 13C CPMAS (SiAl1, ppm) δ 21.9 (CH3C(O) – bidentate), 24.1 (CH3C(O) – monodentate), 33.4, 35.3 (CH3N), 38.2 (CH3N), 170.7 (CH3C(O) – monodentate), 179.4 (CH3C(O) – bidentate); 29Si CPMAS (ppm) δ −89; 27Al MAS (ppm) δ 0 (AlVI = 6-coordinated Al), 29 (AlV = 5-coordinated Al), 54 (AlIV = 4-coordinated Al).
TG/DSC (air, 5 °C min−1) weight loss at 1000 °C 53.83%.
BET: SA = 607 m2 g−1, C = 146, Vtot = 0.35 cm3 g−1, d = 2.4 nm.
Aluminosilicate xerogel C2SiAl with Si2(CH2)2(OAc)6 precursor was prepared with the same procedure and under same conditions as in the case of SiAl1 xerogel. See Table 1.
| Sample | Si precursor | n Si [mmol] | n Al [mmol] | Ala mol% | Template | n template [mmol] | TG residual mass at 1000 °C [%] | TG residual mass calc.b [%] |
|---|---|---|---|---|---|---|---|---|
| a Nominal Almol% = nAl/(nAl + nSi). b Based on AlxSiyOz (see text) in precursors. | ||||||||
| SiAl1 | Si(OAc)4 | 3.907 | 5.018 | 56.2 | — | — | 46.17 | 45.10 |
| C2SiAl | Si2(CH2)2(OAc)6 | 5.108 | 5.100 | 49.9 | — | — | 50.09 | 40.45 |
| SiAl2 | Me3SiOAc | 13.726 | 4.478 | 24.6 | — | — | 7.71 | — |
| SiAlP1 | Si(OAc)4 | 5.029 | 3.460 | 40.8 | P123 | 0.239 | 19.92 | 18.74 |
| SiAlF1 | Si(OAc)4 | 5.025 | 3.416 | 40.5 | F127 | 0.111 | 19.36 | 15.70 |
| SiAlP2 | Si(OAc)4 | 3.825 | 5.206 | 57.6 | P123 | 0.128 | 31.78 | 26.64 |
| SiAlF2 | Si(OAc)4 | 3.894 | 5.114 | 56.8 | F127 | 0.064 | 29.94 | 29.36 |
| SiAlP3 | Si2O(OAc)6 | 2.312 | 2.418 | 51.1 | P123 | 0.071 | 30.49 | 35.59 |
| SiAlF3 | Si2O(OAc)6 | 5.236 | 5.112 | 49.4 | F127 | 0.068 | 31.78 | 39.34 |
Aluminosilicate xerogel prepared with Pluronic F127 template SiAlF2 (eqn (1))
Al(NMe2)3 (0.814 g; 5.11 mmol) dissolved in 25 cm3 of the dry toluene was added dropwise by a syringe to a stirred solution of Si(OAc)4 (1.029 g; 3.894 mmol) and Pluronic F127 (0.800 g) in dry toluene (25 cm3) at 80 °C. After the addition, the mixture color changed to light yellow and a gel formed. The reaction mixture was heated to 80 °C for 12 h forming a transparent light yellow stiff gel. After 7 days of aging at this temperature, the reaction was stopped, volatile byproducts were separated in vacuo and the yellow-orange solid product was dried under vacuum for 24 h. Yield 1.689 g, theor. 0.496 g.O), 1586 s (ν COO), 2867 m (ν CH).
NMR: 13C CPMAS (SiAlF2, ppm) δ 26.9 (CH3C(O)), 40.7 (CH3N), 74.1 (CH2O), 79 (CHO), 173.3 (CH3C(O) – monodentate), 184.6 (CH3C(O) – bidentate); 29Si CPMAS (SiAlF2, ppm) δ −87; 27Al MAS (SiAlF2, ppm) δ 2 (AlVI), 32 (AlV), 61 (AlIV).
TG/DSC: (air, 5 °C min−1) weight loss at 1000 °C 70.06%.
Portions of dried xerogel were calcined in a tube furnace under air at 500 and 700 °C for 3 h.
BET: (sample calcined at 500 °C) SA = 477 m2 g−1, C = 54, Vtot = 0.53 cm3 g−1, d = 5.9 nm.
ICP OES (sample calcined at 500 °C) wt%: Al 25.34 ± 0.04, Si 18.98 ± 0.01.
Hydrothermal stability was studied after calcination at 500 °C. The xerogel was kept in refluxing water for 24 hours. Then, water was evaporated and the sample was dried under vacuum at 100 °C. Changes in the xerogel porosity and structure were studied by N2 porosimetry and MAS NMR spectroscopy.
Calcined samples are labeled with a number corresponding to the particular calcination temperature, e.g. SiAlF1-500, water-treated samples are labeled with H, e.g. SiAlF2-500H.
Reactions with the P123 template were carried out under the same conditions as in the case of F127. Aluminosilicate xerogels from Si2O(OAc)6 precursor were prepared according to same protocol as in the case of SiAlF2 sample. For details see the Table 1.
Condensation reaction between Me3SiOAc and Al(NMe2)3
A model condensation reaction between Me3SiOAc and Al(NMe2)3 (3:1) (Table 1) led to a molecular product (1) containing the N,N-dimethylacetimidic acid cation [Me(C)OHNMe2]+ and [Al(OSiMe3)4]− anion (Fig. 1 and 1S†). Dimethylacetamide, hexamethyldisiloxane, and small amount of dimethylamine were observed by GC-MS among the volatiles. The colorless crystalline product sublimed from the dark orange viscous mass on the walls of a Schlenk flask. These crystals (292 mg, 14%) were separated in glove box and analyzed by single crystal X-ray diffraction analysis.
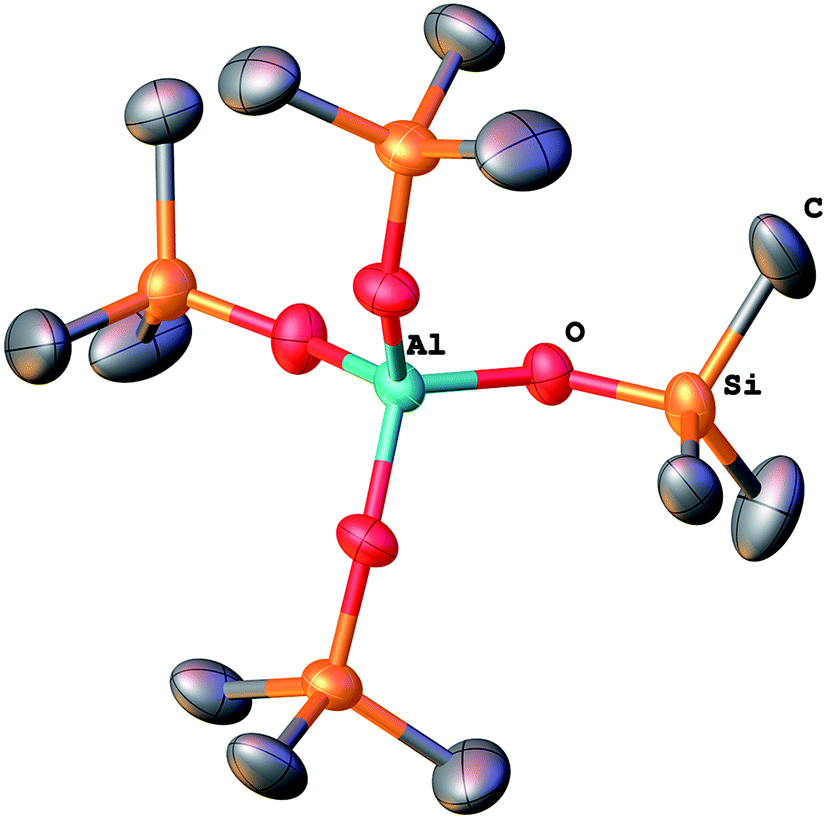 | ||
| Fig. 1 Tetrahedral tetrakis(trimethylsilyloxy)aluminate anion of compound 1. | ||
EI-MS (m/z): 589 [Al2(OSiMe3)6H]+ (10%); 574 [Al2(OSiMe3)5OSiMe2H]+ (100%); 485 [Al2(OSiMe3)4OSiMe2H]+ (20%); 411 [Al2(OSiMe3)4H]+ (50%).
Catalytic reactions
Aminolysis and alcoholysis reactions of styrene oxide33,34,38,44 were performed in a 25 cm3 round-bottomed Schlenk flask connected to a N2 source. Calcined (500 °C) aluminosilicate xerogels were degassed before the reactions under vacuum at 100 °C for 20 min. The reaction mixture consisted of catalyst (25 mg), dry toluene (5 cm3), styrene oxide (0.587 cm3, 5.00 mmol), and aniline (0.456 cm3, 5.00 mmol) or methanol (0.202 cm3, 5.00 mmol) in the case of aminolysis and alcoholysis, respectively. A blank reaction was performed without a catalyst. The reaction mixture was heated at 50 °C. Catalytic products were quantified by GC-MS and 1H NMR spectroscopy after 10 min and 1 h. The reusability of catalyst was studied after washing the used catalyst with CH2Cl2 and toluene and degassing under vacuum at 115 °C. We found that reused catalyst is still active. To investigate Al leaching, 30 mg of the catalyst was refluxed in 15 cm3 of 2-propanol for 4 h. After centrifugation and filtering off the catalyst powder, the filtrate was tested for the presence of Al species by ICP-OES.Results and discussion
Aluminosilicate xerogels were synthesized by a novel acetamide elimination reaction according to eqn (1)–(3) from Si(OAc)4 (or bridged acetoxysilanes) and Al(NMe2)3 precursors in dry toluene under N2 atmosphere.| 3Si(OAc)4 + 4Al(NMe2)3 → Al4Si3O12 + 12Me2NC(O)CH3 | (1) |
| (AcO)3SiOSi(OAc)3 + 2Al(NMe2)3 → Al2Si2O7 + 6Me2NC(O)CH3 | (2) |
| (AcO)3Si–(CH2)2–Si(OAc)3 + 2Al(NMe2)3 → Al2Si2(CH2)2O6 + 6Me2NC(O)CH3 | (3) |
The reaction proceeds with the formation of Si–O–Al network and release of dimethylacetamide which was confirmed by GC-MS (Fig. 2S†) and 1H NMR analysis of volatiles separated from the reaction mixture. In the case of the reaction with excess Si(OAc)4, a small amount of acetic acid was observed in GC-MS.
Hexaacetoxydisiloxane (AcO)3SiOSi(OAc)3 (eqn (2)) and bis(triacetoxysilyl)ethane (AcO)3Si–(CH2)2–Si(OAc)3 (eqn (3)) were used for the modification of porosity and optimizing of the Si/Al ratio. Eqn (1) represents a complete condensation of functional groups to Si–O–Al network and quantitative elimination of dimethylacetamide. However, under experimental conditions, the condensation is incomplete (Scheme 2), and degree of condensation (DC) for the reaction without a template was 56%.
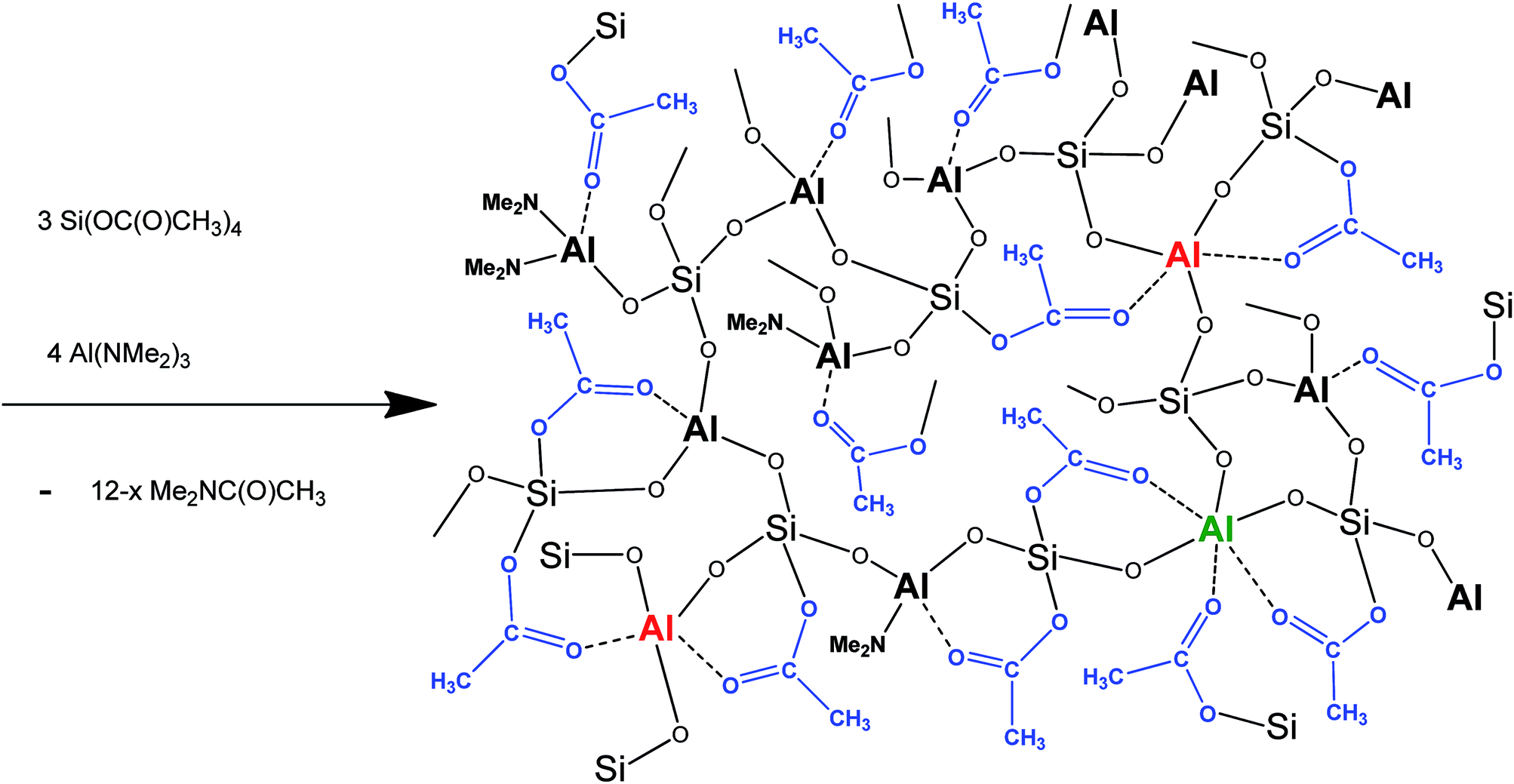 | ||
| Scheme 2 Synthesis of aluminosilicate gel with four-, five-, and six-coordinate Al atoms and residual acetate and dimethylamide groups. | ||
Reaction parameters are summarized in Table 1. Systems without the templates produced yellowish turbid gels which were dried under vacuum for 24 h to form yellow powders. With the use of templates, stiff and transparent yellow gels were obtained. These gels were dried to form yellow-orange rubbery or glassy products. Dried xerogels were afterwards calcined in a tube furnace at 500 °C for 3 h in air to remove the templates. The resulting yellowish powders possessed surface areas that were substantially improved and pore sizes corresponded to mesoporous region (see below).
To study the condensation, a model reaction between Me3SiOAc and Al(NMe2)3 was carried out. It provided molecular ionic compound [Me(C)OHNMe2]+ [Al(OSiMe3)4]− (1) (Fig. 1 and 1S†). Crystallographic details and IR spectrum of 1 are summarized in ESI (Table 1S, Fig. 3S†). A similar compound has been prepared by Chisholm from Me3SiOH and Al(NMe2)3.45 This product shows that condensation between acetoxy and dimethylamido groups takes place forming Si–O–Al bonds.
IR spectra of all dried products were recorded to identify Si–O–Al heterolinkages and also the residual organic groups. Fig. 2 illustrates IR spectra of non-templated and F127 templated dried samples synthesized by NHSG reactions. The absence of OH bands attests to the truly non-aqueous nature of the xerogel synthesis. Vibrational bands located at 1030–1000 cm−1 are attributed to Si–O–Si and Si–O–Al bonds on the basis that introduction of Si–O–Al bonds in a SiO2 matrix shifts the absorption maxima toward the lower wavenumbers.46–48 Besides the bonds mentioned above, vibrational bands of residual organic groups were observed as well. Dimethylamido groups give rise to vibrational bands at 1250–1270, 1370–1380, 1510–1520, and 2970–2980 cm−1.49–51 Acetoxy moieties are characterized by the asymmetric and symmetric COO stretches at 1580–1590 and 1460–1470 cm−1, respectively. The difference between symmetric and asymmetric carboxylate vibrational bands is 120–130 cm−1 and according to Deacon–Phillips rules, this is indicative of the bidentate bridging mode on a metal center.52,53 Pluronic templates P123 and F127 are represented by vibrational bands of C–O–C bonds at 1090–1105 cm−1 and CH3 at 2860–2880 cm−1.54
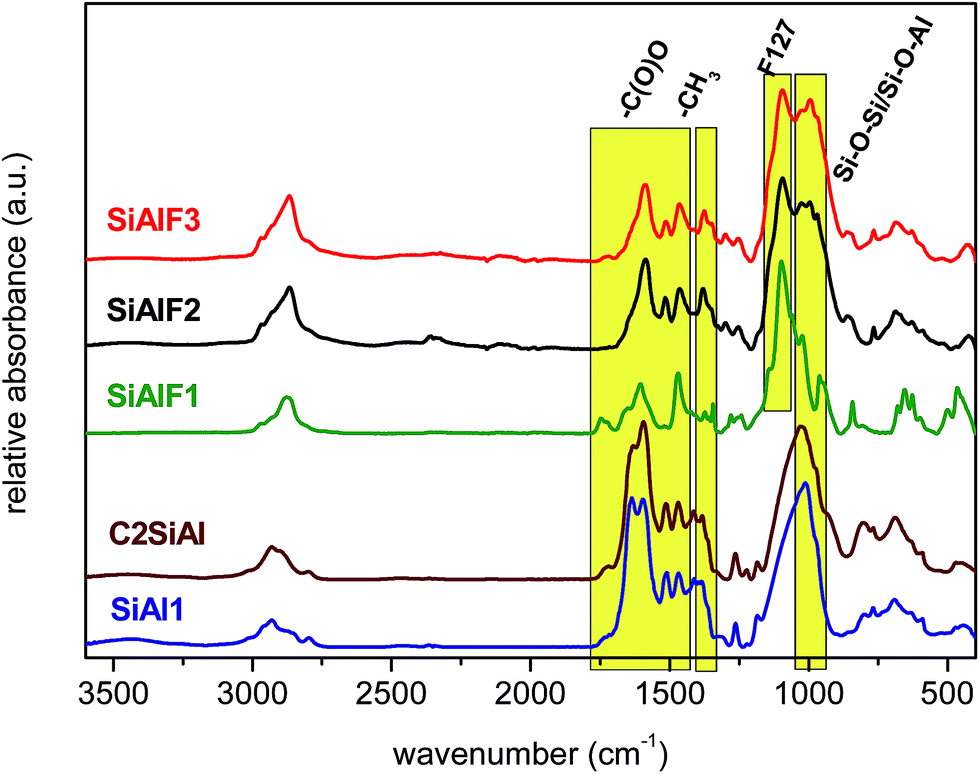 | ||
| Fig. 2 IR spectra of dried aluminosilicate xerogels prepared with and without F127 template. | ||
The thermal behavior of the prepared products was studied by TG/DSC analysis in air. Residual masses are summarized in Table 1. These values are in an agreement with the calculated residual masses corresponding to aluminosilicate AlxSiyOz, where x, y, and z represent moles of aluminium, silicon, and oxygen in precursors. For example, the TG residual mass of sample SiAlF2 (Table 1) is 29.94% while the calculated residual mass of aluminosilicate is 29.36%. TG curves (Fig. 4S†) of xerogels synthesized without the templates display mass losses in two steps. The first mass loss is observed between 50–225 °C (21–26%) and the second one between 230–500 °C (24%). These steps are connected with oxidation of residual dimethylamido and acetoxy moieties. In the case of templated samples the highest mass losses (52–56%) are observed between 200–450 °C. Xerogels with a lower content of Al show the highest mass loss between 125–400 °C (68–72%). A lower decomposition temperature than in the previous case could be explained by the oxidation of unreacted monodentate acetoxy groups bound to silicon. DSC curves of the templated samples (SiAlP2, SiAlF2) with 57–58 mol% of Al show exothermic peaks at 995 and 985 °C for the xerogel with F127 and P123 copolymer, respectively. This observation could be connected to crystallization of the mullite phase.46–48
Solid state NMR spectroscopy (Table 2) was employed for in-depth characterization of the aluminosilicate xerogel matrix. 13C CPMAS NMR spectrum of non-templated sample (SiAl1) displayed resonances of residual organic groups (Fig. 5S†). Resonances at 33.1, 35.0, and 37.8 ppm are attributed to dimethylamido groups while the acetoxy groups in bidentate and monodentate coordination are characterized by the CH3 resonances at 22.0 and 24.1 ppm, respectively. The COO resonances of acetate groups in unidentate and bidentate mode were observed at 170.4 and 179.5 ppm, respectively. Similar resonances of residual N(CH3)2 (34.1, 37.4 ppm), CH3 (20.7, 23.1 ppm) and COO (169.8, 179.0 ppm) were observed also in the 13C CPMAS spectrum of the xerogel synthesized from bis(triacetoxysilyl)ethane (C2SiAl). The ethylene (CH2–CH2) bridge is characterized by the signal at 7.5 ppm. In the 13C CPMAS NMR spectrum of F127 templated xerogel (SiAlF2, Fig. 5S†), the presence of residual organic moieties was observed as well. The N(CH3)2 groups are seen at 40.8 ppm and CH3 of acetoxy species are displayed at 27.3 ppm. The carboxylate function features weak signals at 173.2 and 183.2 ppm. The template carbons are represented by chemical shifts at 21.0 (CH3CHO), 74.1, 76.8, and 78.8 ppm (CH2O and CH3CHO).55
| Sample | Chemical shift [ppm] | |||||
|---|---|---|---|---|---|---|
| 29Si CPMAS | 27Al MAS | |||||
| SiAl1 | −89 | 53 | 28 | 0 | ||
| C2SiAl | −54 | 54 | 29 | −1 | ||
| SiAlF1 | −106 | −97 | 54 | −2 | −4 | |
| SiAlF1-500 | −97 | 61 | 32 | 2 | ||
| SiAlF2 | −88 | 61 | 32 | 2 | ||
| SiAlF2-500 | −90 | 64 | 33 | 4 | ||
| SiAlF3 | −90 | 61 | 30 | 0 | ||
| SiAlF3-500 | −93 | 69 | 34 | 4 | ||
The 29Si CPMAS NMR spectrum of non-templated sample (SiAl1) shows a broad signal with the maximum at −89 ppm (Fig. 3). Compared to SiO2 (−110 ppm)56,57 and Si(OAc)4 (−96 ppm) a downfield shift is observed. This shift is caused by the presence of Si–O–Al linkages in the xerogel network.58–6029Si CPMAS NMR spectrum of the sample C2SiAl with ethylene (CH2–CH2) bridges displays a broad signal with the maximum at −54 ppm (Fig. 3). 29Si CPMAS NMR data of templated samples displays a broad signal at −87 ppm assigned to Si(OSi)(OAl)3 species (Fig. 3).58,59 Similar resonances were observed after calcination at 500 °C of these templated samples (Fig. 4). The resonance at −90 ppm in 29Si CPMAS NMR corresponds to Si(OSi)(OAl)358,59 In the case of templated samples with a lower Al content (SiAlF1), 29Si CPMAS displays a signal with maximum at −106 ppm and a shoulder at −97 ppm. This shift could be caused by the formation of Si–O–Si bonds because of excess Si(OAc)4 (Fig. 3). Comparison of 29Si CPMAS NMR spectra before and after calcinations for the templated samples SiAlF1, 2, 3 indicates there are not major differences (Fig. 4).
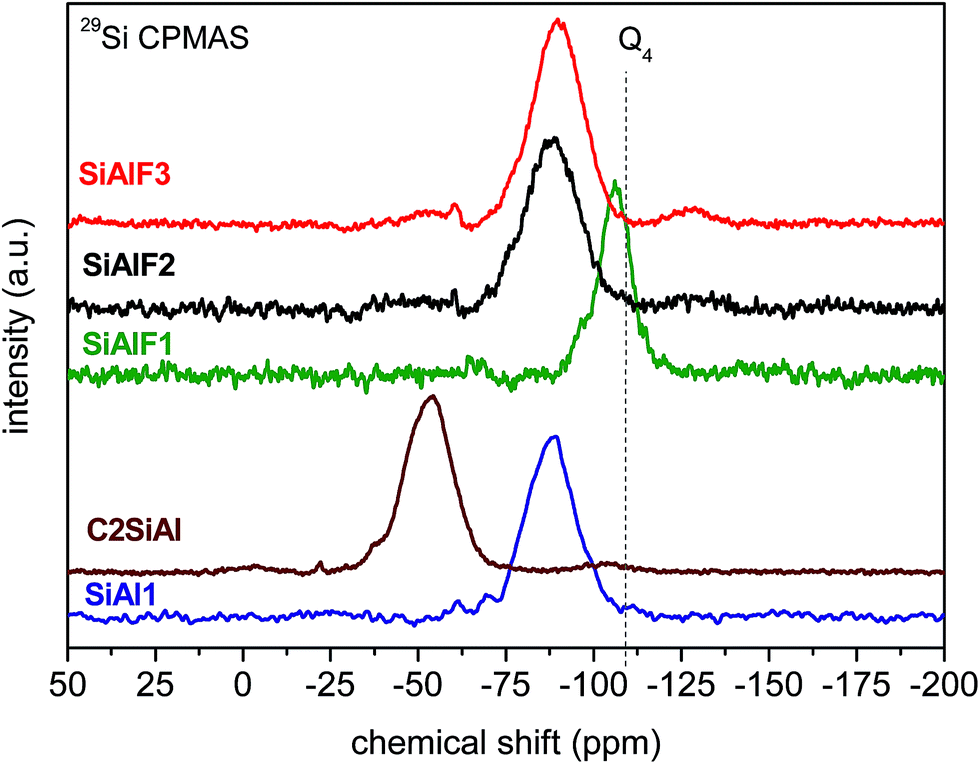 | ||
| Fig. 3 29Si CPMAS NMR spectra of dried aluminosilicate xerogels. | ||
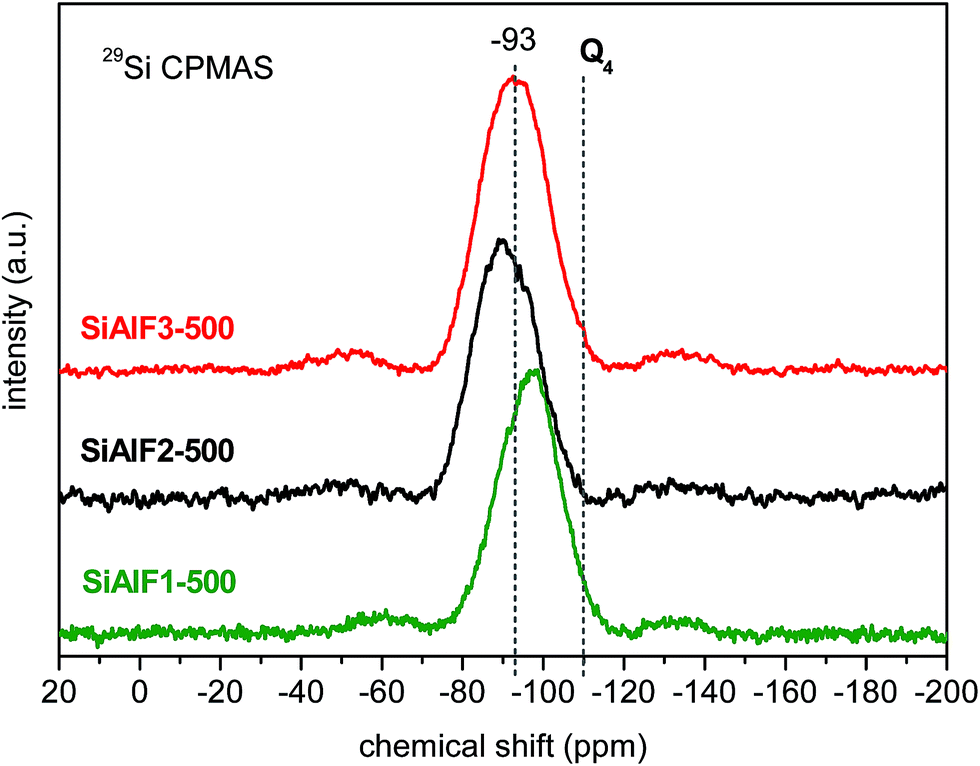 | ||
| Fig. 4 29Si CPMAS NMR spectra of calcined (500 °C) aluminosilicate xerogels synthesized with F127 template. | ||
27Al NMR spectra of dried xerogels provide information about the coordination number of Al atoms in the aluminosilicate network. 27Al spectra of dried non-templated xerogels (SiAl1, C2SiAl) illustrate three signals attributed to 4-coordinated (AlIV, 53 ppm), 5-coordinated (AlV, 28 ppm) and 6-coordinated (AlVI, 0 ppm) Al3,58,61 (Fig. 6S†). 27Al MAS NMR spectra of dried templated samples (SiAlF2, 3) show also the resonances for AlIV (58 ppm), AlV (31 ppm), and AlVI (2 ppm) atoms in the framework (Fig. 6S†). No significant change in the 27Al MAS NMR spectra of calcined templated xerogels was observed and Al signals were found at 62 ppm (AlIV), 33 ppm (AlV), and 4 ppm (AlVI).5827Al MAS NMR spectrum of the sample SiAlF1 (lower Al content) displays signals for AlIV at 54 ppm and for AlVI at −2 and −4 ppm. After calcination of this sample, the resonances at 61 ppm (AlIV), 32 ppm (AlV), and 2 ppm (AlVI) were observed (Fig. 5). Chemical shifts of xerogel samples are summarized in Table 2. From these NMR data we can conclude, that the calcined xerogels still contain Si–O–Al bonds and no phase separation takes place during the calcination. The 27Al NMR and EPR observations made by Chen et al. show association of five-coordinated aluminum atoms to Lewis acid sites.62 Hence, the effect of different aluminum coordination and their acid–base properties on the catalytic activity is a very interesting subject. 27Al MAS spectrum of hydrothermally treated sample SiAlF2-500H shows intensity decrease of signal corresponding to 5-coordinated Al while the signal of 6-coordinated Al atoms increased (Fig. 7S†). This change is caused by coordination of water and –OH groups. Solid state MAS NMR spectra of the samples synthesized with the P123 template are similar and thus are not shown here.
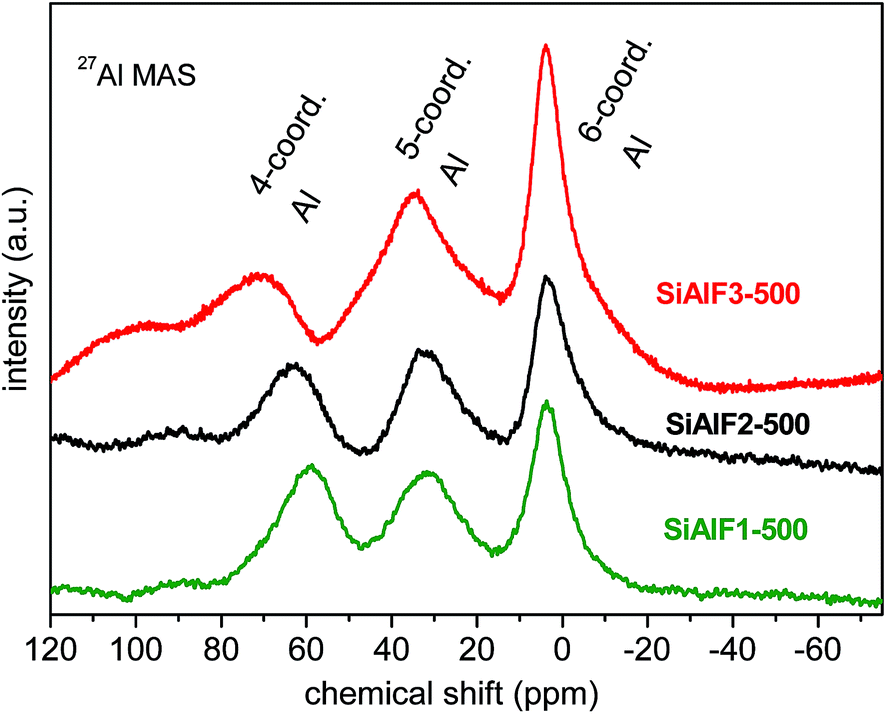 | ||
| Fig. 5 27Al MAS NMR spectra of calcined (500 °C) aluminosilicate xerogels synthesized with F127 template. | ||
A very important feature of these aluminosilicate xerogels is their porosity. High surface area materials with mesopores provide better catalytic efficiency for the processing of bulky organic molecules. We investigated the N2 adsorption/desorption isotherms of the non-templated and templated samples (Table 3). The results show that xerogels synthesized without copolymer templates exhibit high surface areas after drying and feature the type-II isotherms63 (Fig. 6). Equivalent BET surface areas of xerogels prepared from Si(OAc)4 (SiAl1) and from the ethylene bridged acetoxysilane (C2SiAl, Fig. 8S†) were 607 and 517 m2 g−1, respectively. However the pore diameters correspond to micropores in both materials. After calcination of the C2SiAl sample at 200 °C, surface area decreased considerably to 36 m2 g−1. This change can be explained by the framework collapse while the ethylene bridging groups are oxidized. Interestingly, according to the NLDFT method, the pore diameter increased and shifted to the mesopore region (4.8 nm).
| Sample | Template | Al mol% | SA (BET) [m2 g−1] | V [cm3 g−1] | d [nm] |
|---|---|---|---|---|---|
| SiAl1 | — | 56.2 | 607 | 0.34 | 2.4 |
| C2SiAl | — | 49.9 | 517 | 0.38 | 2.9 |
| SiAlP1-500 | P123 | 42.2 | 429 | 0.37 | 4.9 |
| SiAlF1-500 | F127 | 40.6 | 381 | 0.37 | 4.9 |
| SiAlP2-500 | P123 | 59.3 | 591 | 0.53 | 4.9 |
| SiAlF2-500 | F127 | 58.1 | 477 | 0.53 | 5.9 |
| SiAlF2-700 | F127 | 58.1 | 424 | 0.41 | 5.1 |
| SiAlF2-500H | F127 | 58.1 | 166 | 0.28 | 6.1 |
| SiAlP3-500 | P123 | 54.1 | 627 | 0.55 | 4.9 |
| SiAlF3-500 | F127 | 50.7 | 442 | 0.46 | 5.3 |
| SiAlF3-500H | F127 | 50.7 | 171 | 0.31 | 6.1 |
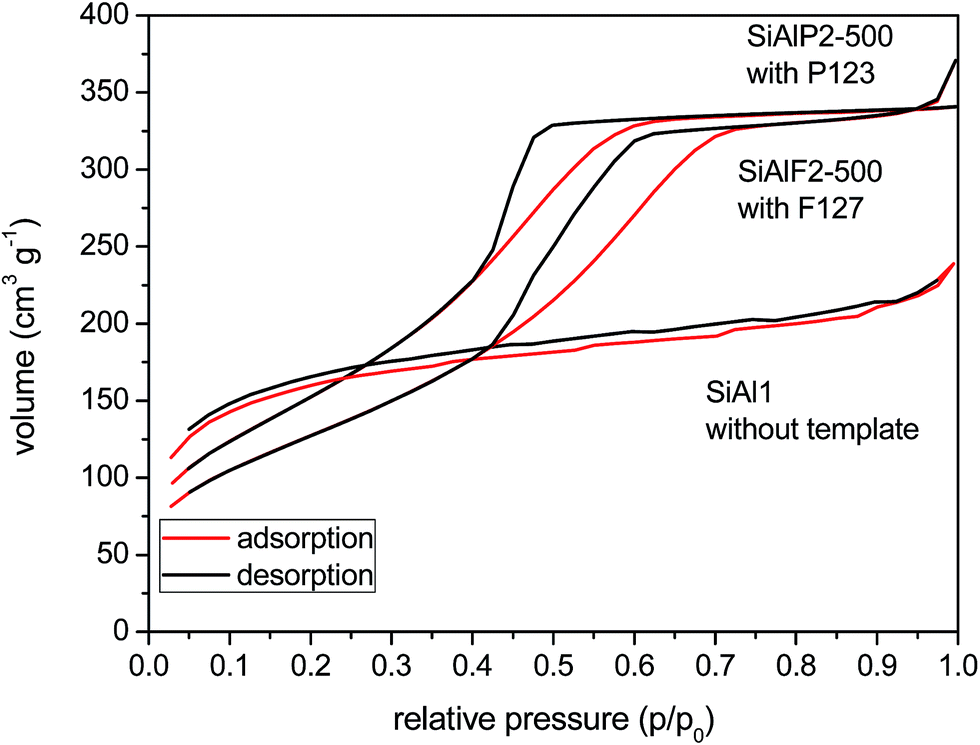 | ||
| Fig. 6 N2 adsorption/desorption isotherms of aluminosilicate xerogels. | ||
An improvement in morphological properties is achieved through the use of Pluronic templates (Table 3). With this approach, framework collapse during heat treatment is avoided and high surface areas are achieved after calcination at 500 °C. The shape of isotherms corresponds to type IV with the H2 hysteresis consistent with significant mesopore content and narrower pore size distributions (Fig. 6 and 9S†).63 Calcined xerogel SiAlP2-500 synthesized with the P123 template containing 59.3 mol% of Al exhibits surface area of 591 m2 g−1 and the pore volume 0.53 cm3 g−1. If (AcO)3SiOSi(OAc)3 is used as the silicon precursor, the surface area increases to 627 m2 g−1. This change can be caused by better crosslinking due to a higher number of functional groups. Pore diameters of these calcined samples calculated by the NLDFT method are between 4.9 and 5.9 nm (Table 3). In the case of the aluminosilicates synthesized with the F127 template, surface areas in the range 381–477 m2 g−1 are achieved with a pore diameter of 4.9–5.9 nm (Fig. 10S†). We observed that with increasing Al content the surface areas and pore volumes increased as well. Pore diameters do not change with different Si/Al ratios for P123 but increased for F127. For classification of thermal stability, we calcined the sample SiAlF2 at 700 °C for 3 h and remeasured N2 adsorption isotherms (Fig. 7). Its surface area reached 424 m2 g−1 (Vtot = 0.41 cm3 g−1) which is about 53 m2 g−1 lower than in SiAlF2-500 and the pore volume and diameter decreased (Fig. 8, Table 3) and the distribution is narrower. To investigate hydrothermal stability, the sample SiAlF2-500 was exposed to boiling water for 24 h (Fig. 7). The surface area after this test was reduced to 166 m2 g−1 and pore volume to 0.28 cm3 g−1. The pore size distribution maximum moved to 6.1 nm and the mesoporous character was preserved (Fig. 8). Compared to this sample, the hydrothermally treated sample (SiAlF3-500H) synthesized with (AcO)3SiOSi(OAc)3 exhibits also loss of surface area and pore volume while pore diameter increased (Table 3, Fig. 11S†).
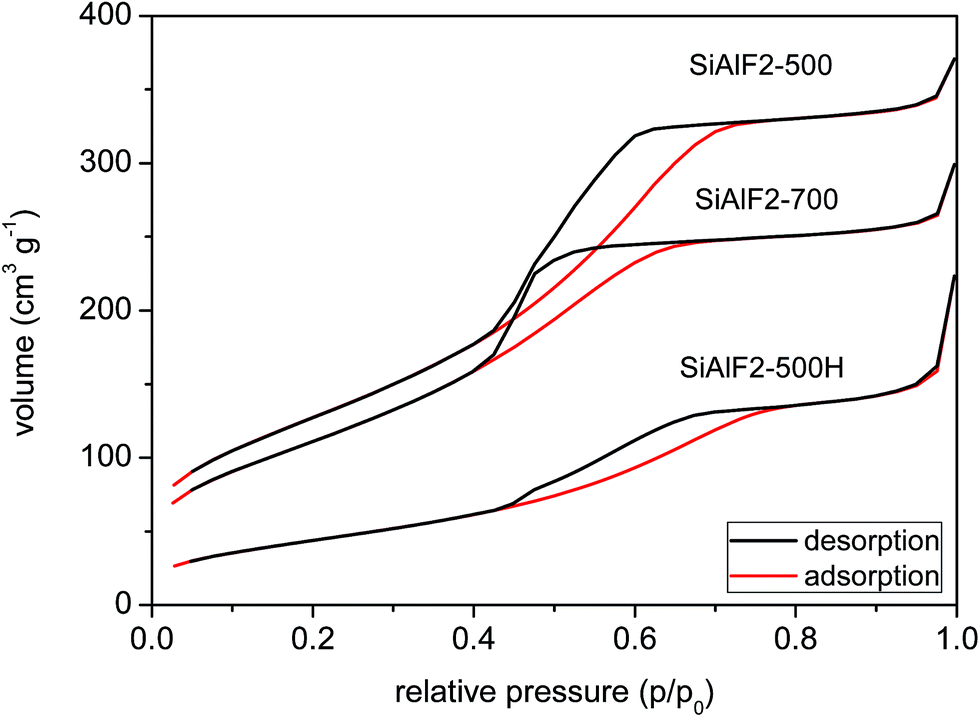 | ||
| Fig. 7 N2 adsorption/desorption isotherms of aluminosilicate xerogel SiAlF2 calcined at 500 (top) and 700 °C (middle). Sample SiAlF2-500H after hydrothermal treatment (bottom). | ||
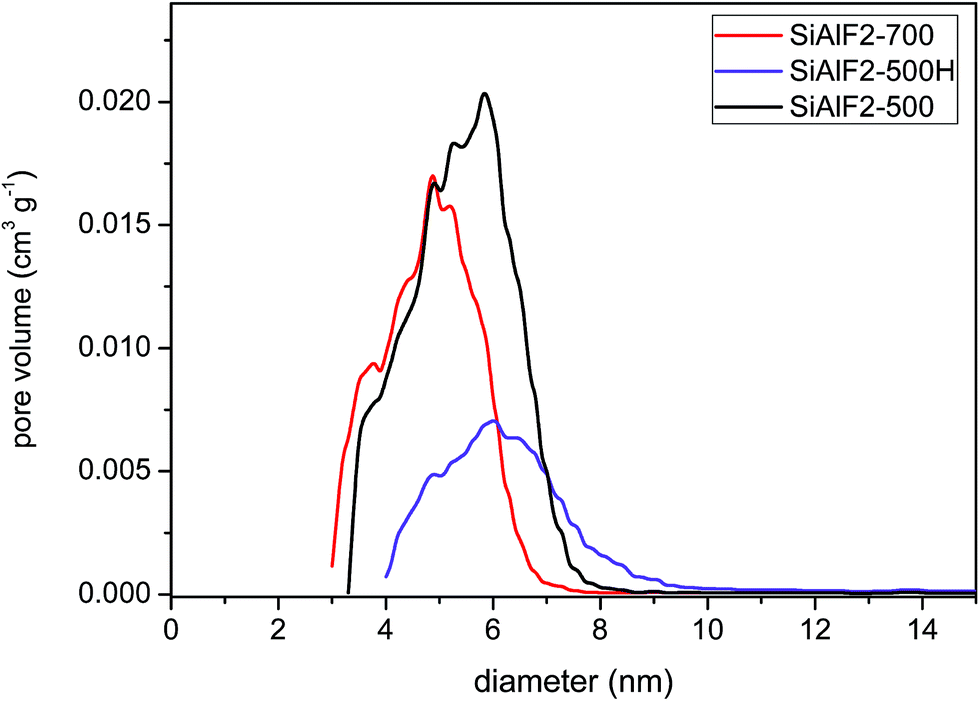 | ||
| Fig. 8 NLDFT pore diameters (adsorption branch) of aluminosilicate xerogels: SiAlF2 calcined at 500 and 700 °C. Sample SiAlF2-500H after hydrothermal treatment. | ||
SAXS measurements of the calcined samples SiAlP2-500 and SiAlF2-500 synthesized with the P123 and F127, respectively, show a weak scattering maximum at 0.42° (0.06 Å−1) (Fig. 9). This behavior points to the mesoscopic ordering with the uniform pore channels in the xerogel.
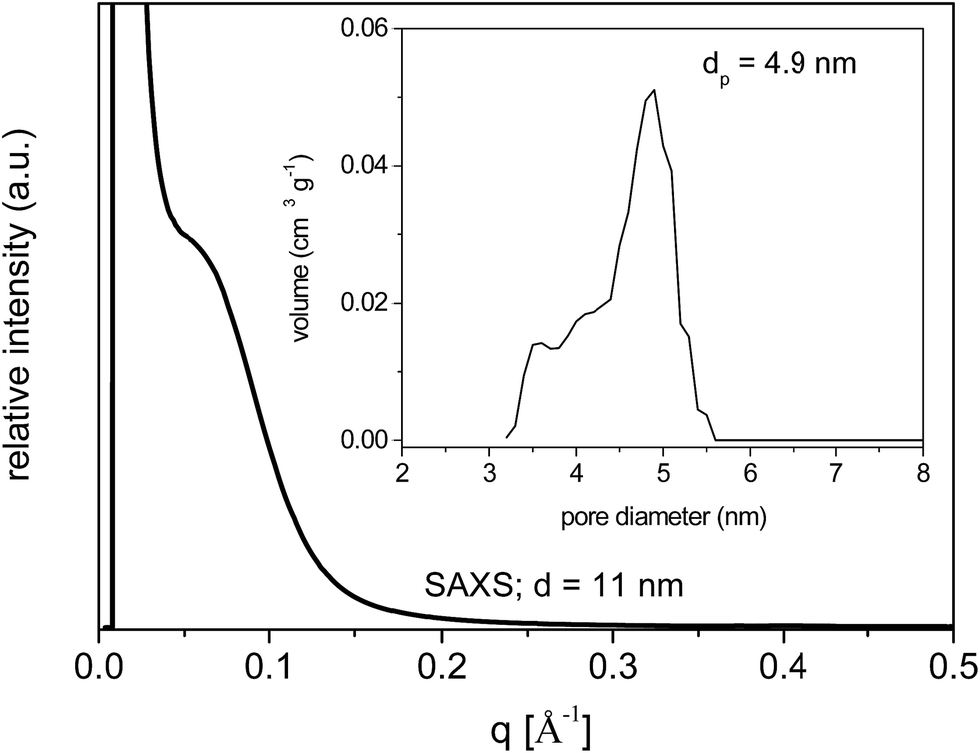 | ||
| Fig. 9 SAXS pattern of calcined aluminosilicate xerogel SiAlP2-500. NLDFT pore diameter distribution (inset). | ||
The morphology of calcined mesoporous xerogels was studied by the TEM technique. The micrographs illustrate “wormhole” structure of pores (Fig. 10).
 | ||
| Fig. 10 TEM micrographs of SiAlF3-500 aluminosilicate with mesoporous “wormhole” like morphology. | ||
The surface acidity of the calcined xerogels was studied by pyridine adsorption. The IR spectra of the pyridine-treated samples (Fig. 11) display weak absorption bands at 1450, 1492, 1548, 1599, 1625, and 1639 cm−1. The bands corresponding to Lewis acid sites (LPy) are present at 1450 and 1625 cm−1.61,64 Both Lewis and Brønsted acid sites are attributed to a vibrational band at 1492 cm−1. Brønsted acidic sites (BPy) are characterized by the absorption bands at 1548 and 1639 cm−1.64 Pyridine adsorbed by hydrogen bonds (HPy) of ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–OH moieties can be assigned to the vibrational band at 1599 cm−1.61 These findings show that synthesized aluminosilicate xerogels exhibit surface acidity of both Lewis and Brønsted type.
Si–OH moieties can be assigned to the vibrational band at 1599 cm−1.61 These findings show that synthesized aluminosilicate xerogels exhibit surface acidity of both Lewis and Brønsted type.
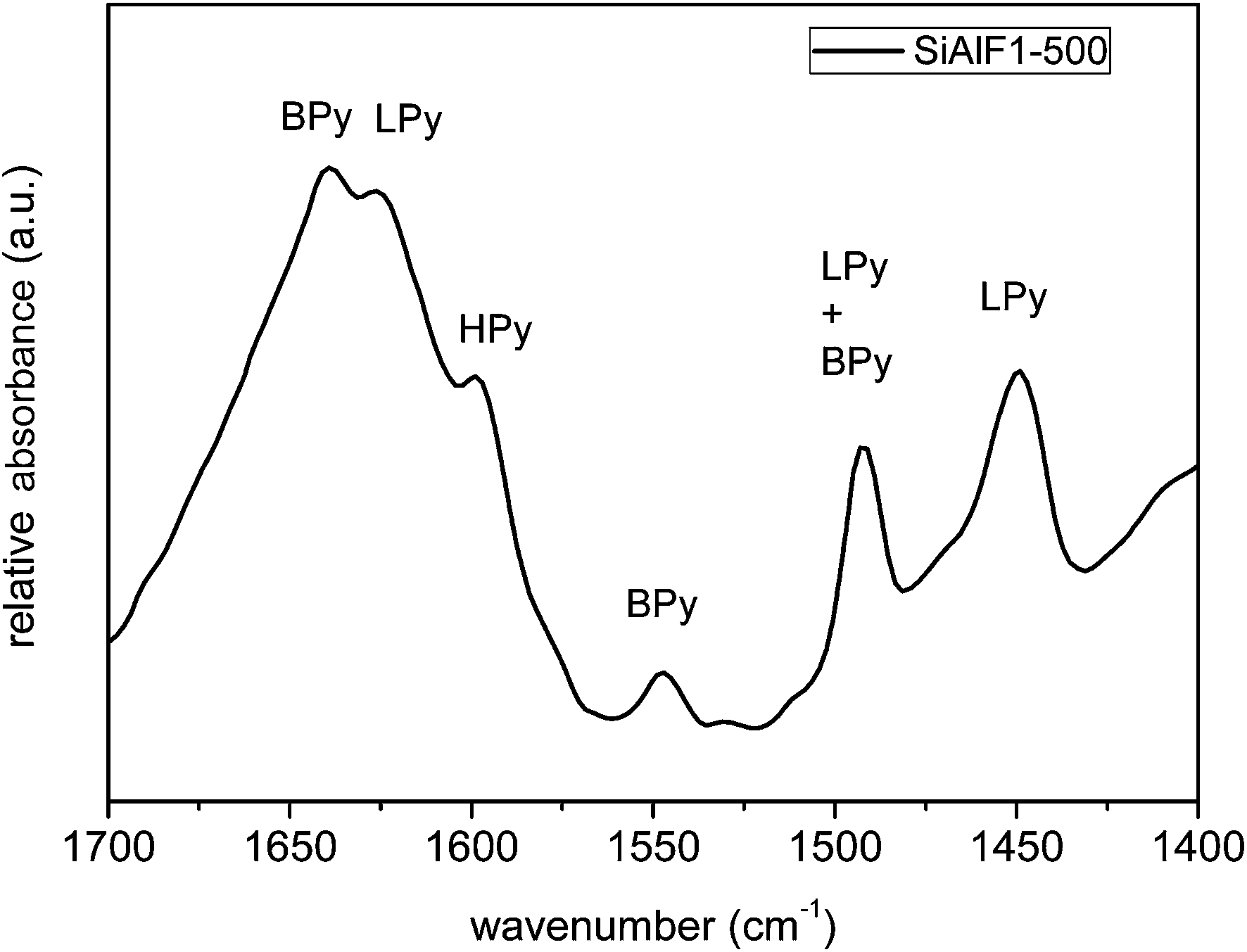 | ||
| Fig. 11 IR spectra of calcined aluminosilicate xerogel (SiAlF1-500) after pyridine adsorption. | ||
High-temperature powder X-ray diffraction (HT PXRD) was used to determine the crystal phases that arise during calcination (Fig. 12S†). It was found that templated samples remain amorphous up to high temperatures and then crystallize to mullite (PDF 82-1237).65–67 The xerogels SiAlP2 and SiAlF2 are amorphous up to 950 °C before the diffraction lines of mullite are observed at 1000 °C (Fig. 12S†). These findings correspond to exothermic effects in DSC analysis (995 and 985 °C). In the case of xerogels with a lower content of Al, diffraction evidence for mullite appeared at 1050 °C. Besides the mullite phase, crystalline SiO2 (cristobalite PDF 85-0621) at 1100 °C was observed as well. The presence of cristobalite could be explained by the excess of Si(OAc)4 in the starting mixture which results in the phase separation during calcination. Samples synthesized with (AcO)3SiOSi(OAc)3 (SiAlF3-500 and SiAlP3-500) exhibit the mullite phase with crystallization temperature between 950–1000 °C. Crystallization of mullite at ∼980 °C indicates the homogeneity of aluminosilicate gels at the atomic level.26,68,69 These findings show that our mesoporous aluminosilicates can be transformed to homogeneous mullite phase.
Catalytic studies
Catalytic activities of the synthesized xerogels were tested in two types of model reactions (Fig. 13S and 14S†). At first, aminolysis of styrene oxide with aniline34,44 was investigated with the calcined aluminosilicate xerogel as a catalyst. The second model reaction was alcoholysis of styrene oxide with methanol.33 Catalytic yields for aminolysis are summarized in Table 4. The blank reaction without a catalyst was performed as well and no product was obtained after 4 h. We observed that our calcined mesoporous xerogels are efficient catalysts for aminolysis reaction of epoxides. The 1H NMR spectra of the reaction mixture (in C6D6) display resonances attributed to two products: 2-phenyl-2-(phenylamino)ethanol (Ia) and 1-phenyl-2-(phenylamino)ethanol (IIa) (Fig. 13S and 15S†). These products were also confirmed by the GC-MS technique. Catalytic yields were analyzed after 10 and 60 minutes. The highest conversion was observed with the catalyst SiAlF1-500 (82% after 1 hour). However, high catalytic efficiencies (up to 80%) were reached in all cases (Table 4). Catalyst with a high content of Al exhibits a lower catalytic activity which could be caused by the presence of larger alumina clusters. Xerogels synthesized with (AcO)3SiOSi(OAc)3 provide higher homogeneity and better dispersion of Al in framework and furthermore a higher catalytic activity. We also investigated recycling the catalysts in the second reaction run. The styrene oxide conversion with 7 mg of reused xerogel (SiAlF3-500) reached 21% after 1 h which represents nearly the same activity (TOF = 20) as the fresh catalyst. A report describing aluminosilicates as catalysts for aminolysis was published by Robinson.70 These nanoporous catalysts are prepared by conventional hydrolytic sol–gel process from TEOS and aluminium nitrate nonahydrate in the presence of cetyltrimethylammonium bromide. The comparison between these catalysts and our mesoporous aluminosilicates is presented in Table 5.| Sample | Al mol% (ICP) | n Al in catal. (25 mg) [mmol] | Time [min] | Conversion [%] | Selectivity [%] | TOFa | |
|---|---|---|---|---|---|---|---|
| Ia | IIa | ||||||
| a TOF – apparent turnover frequency [mmol mmol−1 h−1]. | |||||||
| SiAlP1-500 | 42.2 | 0.16 | 10 | 46 | 96 | 4 | 86 |
| 60 | 79 | 95 | 5 | 25 | |||
| SiAlF1-500 | 40.6 | 0.15 | 10 | 50 | 97 | 3 | 100 |
| 60 | 82 | 98 | 2 | 27 | |||
| SiAlF2-500 | 58.1 | 0.23 | 10 | 43 | 96 | 4 | 56 |
| 60 | 69 | 95 | 5 | 15 | |||
| SiAlF3-500 | 50.7 | 0.19 | 10 | 50 | 96 | 4 | 79 |
| 60 | 79 | 96 | 4 | 21 | |||
| SiAlP3-500 | 54.1 | 0.21 | 10 | 56 | 96 | 4 | 80 |
| 60 | 80 | 96 | 4 | 19 | |||
| Sample | Si/Al | Temperature [°C] | Time [h] | m [mg] | Conversion [%] | Selectivity (Ia:IIa) [%] |
|---|---|---|---|---|---|---|
| SiAlF1-500 | 1.5 | 50 | 1 | 25 | 82 | 98:2 |
| SiAlF2-500 | 0.7 | 50 | 1 | 25 | 69 | 95:5 |
| SiAlF3-500 | 1.0 | 50 | 1 | 25 | 80 | 96:4 |
| AS-(14)70 | 14 | r. t. | 6 | 50 | 37 | 95:5 |
| AS-(14)70 | 14 | r. t. | 24 | 50 | 53 | 95:5 |
| AS-(14)70 | 14 | 40 | 6 | 50 | 28 | 95:5 |
| AlKIT-5(10)38 | 10 | r. t. | 0.5 | 50 | 87 | — |
In the case of styrene oxide alcoholysis, our catalysts exhibit a lower efficiency than in aminolysis reactions. Catalytic products confirmed by GC-MS and 1H NMR were (Fig. 15S and 16S†) (1,2-dimethoxyethyl)-benzene (Ib) and phenylacetaldehyde (IIb) (Fig. 14S,†Table 6). Catalyst SiAlF1-500 exhibits 28 and 44% conversion of styrene oxide after 2 and 5 h, respectively. Selectivity for (1,2-dimethoxyethyl)-benzene after 5 h reached 54%.
| Sample | Al mol% (ICP) | n Al in catal. [mmol] | Time [h] | Conversion [%] | Selectivity [%] | |
|---|---|---|---|---|---|---|
| Ib | IIb | |||||
| SiAlF1-500 | 40.6 | 0.15 | 2 | 28 | 63 | 37 |
| 3 | 32 | 59 | 41 | |||
| 5 | 44 | 54 | 46 | |||
We also performed a catalytic reaction with the catalyst SiAlF1-500 where no methanol was used. In this case, phenylacetaldehyde was observed as the only product and the catalytic yield after 1 h was 35%. A leaching test confirmed that no Al was released into the solution. Only 0.3% of the total aluminium amount in catalyst was found in the filtrate.
Conclusions
The non-hydrolytic sol–gel (NHSG) reactions based on acetamide elimination described here provide a very effective route to homogeneous aluminosilicate xerogels with high Al loading. This one pot reaction proceeds as polycondensation between acetoxysilanes and aluminium(III) dimethylamide and leads to the formation of Al–O–Si networks. In all cases N,N-dimethylacetamide was confirmed as the only reaction byproduct attesting to the absence of homocondensation reactions. Al–O–Si bonds were identified by IR spectroscopy and 29Si and 27Al MAS NMR spectroscopy. We observed 4-, 5-, and 6-coordinated Al atoms in the frameworks of both dried and calcined xerogels. These materials exhibit high surface areas (up to 607 m2 g−1) but are microporous. Calcination causes framework collapse and samples lose porosity. With the use of block copolymer templates such as Pluronic P123 and F127, homogeneous, stiff gels were obtained. After calcination at 500 and 700 °C mesoporous aluminosilicate materials with high surface areas ranging from 380 to 627 m2 g−1 with the pore diameters 4.9 (P123) and 5.9 (F127) nm were obtained. After hydrothermal treatment, the surface area decreased but mesoporous character was preserved. Calcined xerogels were investigated as catalysts for the aminolysis and alcoholysis of styrene oxide. The presence of catalytic active acidic sites (Lewis, Brønsted) was determined by pyridine adsorption. All xerogels exhibit good catalytic activity. Especially in the aminolysis model reaction, very high conversions (69–82%) were observed.Acknowledgements
This research has been financially supported by the Ministry of Education, Youth and Sports of the Czech Republic under the project CEITEC 2020 (LQ1601). Authors thank the project KONTAKT II LH11028, the X-ray Diffraction and Bio-SAXS Core Facility of CEITEC, L. Simonikova for ICP-OES analyses, and V. Vykoukal for the SAXS measurements.References
- D. S. Santilli and S. I. Zones, Catal. Lett., 1990, 7, 383–387 CrossRef CAS.
- G. F. Wan, A. J. Duan, Y. Zhang, Z. Zhao, G. Y. Jiang, D. Q. Zhang, Z. Y. Gao, J. Liu and K. H. Chung, Energy Fuels, 2009, 23, 3846–3852 CrossRef CAS.
- E. Masika and R. Mokaya, Chem. Mater., 2011, 23, 2491–2498 CrossRef CAS.
- G. A. Pozarnsky and A. V. McCormick, J. Non-Cryst. Solids, 1995, 190, 212–225 CrossRef CAS.
- J. Ren, L. Zhang and H. Eckert, J. Sol-Gel Sci. Technol., 2014, 70, 482–490 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- R. Mokaya and W. Jones, Chem. Commun., 1998, 1839–1840 RSC.
- K. Wang, Y. Lin, M. A. Morris and J. D. Holmes, J. Mater. Chem., 2006, 16, 4051 RSC.
- D. Zhao, Q. Huo, J. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS.
- A. Sakthivel, S. J. Huang, W. H. Chen, Z. H. Lan, K. H. Chen, H. P. Lin, C. Y. Mou and S. B. Liu, Adv. Funct. Mater., 2005, 15, 253–258 CrossRef CAS.
- C. Li, Y. Wang, Y. Guo, X. Liu, Y. Guo, Z. Zhang, Y. Wang and G. Lu, Chem. Mater., 2007, 19, 173–178 CrossRef CAS.
- S. Li, Q. Xu, J. Chen and Y. Guo, Ind. Eng. Chem. Res., 2008, 47, 8211–8217 CrossRef CAS.
- H. Liu, L. Wang, W. Feng, L. Cao, X. Gao, H. Liu and C. Xu, Ind. Eng. Chem. Res., 2013, 52, 3618–3627 CAS.
- G. M. Kumaran, S. Garg, K. Soni, M. Kumar, L. D. Sharma, K. S. Rama Rao and G. M. Dhar, Ind. Eng. Chem. Res., 2007, 46, 4747–4754 CrossRef CAS.
- A. Vinu, D. P. Sawant, K. Ariga, M. Hartmann and S. B. Halligudi, Microporous Mesoporous Mater., 2005, 80, 195–203 CrossRef CAS.
- Q. Li, Z. Wu, B. Tu, S. S. Park, C.-S. Ha and D. Zhao, Microporous Mesoporous Mater., 2010, 135, 95–104 CrossRef CAS.
- C. Han, H. Wang, L. Zhang, R. Li, Y. Zhang, Y. Luo and X. Zheng, Adv. Powder Technol., 2011, 22, 20–25 CrossRef CAS.
- Y. Liu, W. Zhang and T. J. Pinnavaia, Angew. Chem., Int. Ed., 2001, 40, 1255–1258 CrossRef CAS.
- Y. Liu and T. J. Pinnavaia, Chem. Mater., 2002, 14, 3–5 CrossRef CAS.
- Z. Zhang, Y. Han, L. Zhu, R. Wang, Y. Yu, S. Qiu, D. Zhao and F.-S. Xiao, Angew. Chem., Int. Ed., 2001, 40, 1258–1262 CrossRef CAS.
- H. Wang, Y. Liu and T. J. Pinnavaia, J. Phys. Chem. B, 2006, 110, 4524–4526 CrossRef CAS PubMed.
- D. Trong On and S. Kaliaguine, Angew. Chem., Int. Ed., 2002, 41, 1036–1040 CrossRef.
- D. Trong On and S. Kaliaguine, J. Am. Chem. Soc., 2003, 125, 618–619 CrossRef PubMed.
- H. Liu, K. Guo, X. Li, S. Liu, X. Gao, H. Liu, L. Cao, Y. Chen and C. Xu, Microporous Mesoporous Mater., 2014, 188, 108–117 CrossRef CAS.
- M. Enterría, F. Suárez-García, A. Martínez-Alonso and J. M. D. Tascón, J. Alloys Compd., 2014, 583, 60–69 CrossRef.
- A. Vioux, Chem. Mater., 1997, 9, 2292–2299 CrossRef.
- S. Acosta, R. Corriu, D. Leclercq, P. H. Mutin and A. Vioux, J. Sol-Gel Sci. Technol., 1994, 2, 25–28 CrossRef CAS.
- D. P. Debecker, K. Bouchmella, M. Stoyanova, U. Rodemerck, E. M. Gaigneaux and P. Hubert Mutin, Catal. Sci. Technol., 2012, 2, 1157–1164 CAS.
- K. Bouchmella, P. Hubert Mutin, M. Stoyanova, C. Poleunis, P. Eloy, U. Rodemerck, E. M. Gaigneaux and D. P. Debecker, J. Catal., 2013, 301, 233–241 CrossRef CAS.
- D. Janackovic, A. Orlovic, D. Skala, S. Drmanic, L. Kostic-Gvozdenovic, V. Jokanovic and D. Uskokovic, Nanostruct. Mater., 1999, 12, 147–150 CrossRef.
- J.-C. Kim, K. Cho, S. Lee and R. Ryoo, Catal. Today, 2015, 243, 103–108 CrossRef CAS.
- P. Neves, S. Lima, M. Pillinger, S. M. Rocha, J. Rocha and A. A. Valente, Catal. Today, 2013, 218–219, 76–84 CrossRef CAS.
- M. W. C. Robinson, R. Buckle, I. Mabbett, G. M. Grant and A. E. Graham, Tetrahedron Lett., 2007, 48, 4723–4725 CrossRef CAS.
- M. W. C. Robinson, D. A. Timms, S. M. Williams and A. E. Graham, Tetrahedron Lett., 2007, 48, 6249–6251 CrossRef CAS.
- A. Styskalik, D. Skoda, J. Pinkas and S. Mathur, J. Sol-Gel Sci. Technol., 2012, 63, 463–472 CrossRef CAS.
- D. Skoda, A. Styskalik, Z. Moravec, P. Bezdicka, C. Barnes and J. Pinkas, J. Sol-Gel Sci. Technol., 2015, 74, 810–822 CrossRef CAS.
- D. Skoda, A. Styskalik, Z. Moravec, P. Bezdicka and J. Pinkas, J. Mater. Sci., 2015, 50, 3371–3382 CAS.
- R. Chakravarti, H. Oveisi, P. Kalita, R. R. Pal, S. B. Halligudi, M. L. Kantam and A. Vinu, Microporous Mesoporous Mater., 2009, 123, 338–344 CrossRef CAS.
- D. Barreca, M. P. Copley, A. E. Graham, J. D. Holmes, M. A. Morris, R. Seraglia, T. R. Spalding and E. Tondello, Appl. Catal., A, 2006, 304, 14–20 CrossRef CAS.
- J. Goubeau and R. Mundiel, Z. Anorg. Allg. Chem., 1953, 272, 313–325 CrossRef CAS.
- J. Rouquerol, F. Rouquerol, P. Llewellyn, G. Maurin and K. S. W. Sing, Adsorption by Powders and Porous Solids: Principles, Methodology and Applications, Elsevier Science, 2013 Search PubMed.
- S. Lowell, J. Shields, M. Thomas and M. Thommes, in Characterization of Porous Solids and Powders: Surface Area, Pore Size and Density, Springer Netherlands, 2004, vol. 16, ch. 5, pp. 58–81 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- R. Kore, R. Srivastava and B. Satpati, ACS Catal., 2013, 3, 2891–2904 CrossRef CAS.
- M. H. Chisholm, J. C. Huffman and J. L. Wesemann, Polyhedron, 1991, 10, 1367–1372 CrossRef CAS.
- Y. Hirata, K. Sakeda, Y. Matsushita, K. Shimada and Y. Ishihara, J. Am. Ceram. Soc., 1989, 72, 995–1002 CrossRef CAS.
- E. Ruiz de Sola, F. Estevan, F. J. Torres and J. Alarcón, J. Non-Cryst. Solids, 2005, 351, 1202–1209 CrossRef CAS.
- C. S. Blackwell, J. Phys. Chem., 1979, 83, 3251–3257 CrossRef CAS.
- A. M. Dwivedi, S. Krimm and S. Mierson, Spectrochim. Acta, Part A, 1989, 45, 271–279 CrossRef.
- G. Socrates, Infrared and Raman characteristic group frequencies : tables and charts, John Wiley & Sons, West Sussex, 2007 Search PubMed.
- V. Venkata Chalapathi and K. Venkata Ramiah, Proc. Indiana Acad. Sci., 1968, 68, 109–122 Search PubMed.
- G. Deacon and R. Phillips, Coord. Chem. Rev., 1980, 33, 227–250 CrossRef CAS.
- S. Doeuff, M. Henry, C. Sanchez and J. Livage, J. Non-Cryst. Solids, 1987, 89, 206–216 CrossRef CAS.
- Y.-l. Su, J. Wang and H.-z. Liu, Langmuir, 2002, 18, 5370–5374 CrossRef CAS.
- H.-M. Kao, C.-L. Chen and S.-W. Chiao, J. Chin. Chem. Soc., 2005, 52, 693–699 CrossRef CAS.
- R. Simonutti, A. Comotti, S. Bracco, A. Simonelli and P. Sozzani, Chem. Mater., 2002, 14, 3377–3381 CrossRef CAS.
- D. W. Sindorf and G. E. Maciel, J. Phys. Chem., 1982, 86, 5208–5219 CrossRef CAS.
- M. Toba, F. Mizukami, S.-i. Niwa, T. Sano, K. Maeda and H. Shoji, J. Mater. Chem., 1994, 4, 1131 RSC.
- E. Lippmaa, M. Maegi, A. Samoson, M. Tarmak and G. Engelhardt, J. Am. Chem. Soc., 1981, 103, 4992–4996 CrossRef CAS.
- I. Jaymes, A. Douy, D. Massiot and J.-P. Coutures, J. Non-Cryst. Solids, 1996, 204, 125–134 CrossRef CAS.
- C. Hernandez and A. C. Pierre, Langmuir, 2000, 16, 530–536 CrossRef CAS.
- F. R. Chen, J. G. Davis and J. J. Fripiat, J. Catal., 1992, 133, 263–278 CrossRef CAS.
- M. Thommes, Chem. Ing. Tech., 2010, 82, 1059–1073 CrossRef CAS.
- M. A. Betiha, M. F. Menoufy, A. M. Al-Sabagh, H. M. A. Hassan and S. A. Mahmoud, Microporous Mesoporous Mater., 2015, 204, 15–24 CrossRef CAS.
- H. Schneider, J. Schreuer and B. Hildmann, J. Eur. Ceram. Soc., 2008, 28, 329–344 CrossRef CAS.
- S. Satoshi, C. Contreras, H. Juárez, A. Aguilera and J. Serrato, Int. J. Inorg. Mater., 2001, 3, 625–632 CrossRef CAS.
- D. M. D. D. R. Treadwell and I. A. Aksay, Chem. Mater., 1996, 8, 2056–2060 CrossRef CAS.
- A. Navrotsky, Chem. Mater., 1994, 6, 160–170 CrossRef.
- F. He and W. T. Petuskey, Mater. Lett., 2009, 63, 2631–2634 CrossRef CAS.
- M. W. C. Robinson, A. M. Davies, I. Mabbett, T. E. Davies, D. C. Apperley, S. H. Taylor and A. E. Graham, J. Mol. Catal. A: Chem., 2010, 329, 57–63 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1437252. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra24563j |
| This journal is © The Royal Society of Chemistry 2016 |