
Magnetic and magnetocaloric properties of iron substituted holmium chromite and dysprosium chromite
Abstract
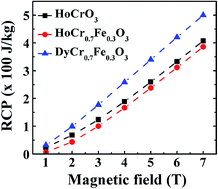
In this work, HoCrO3 and Fe substituted HoCrO3 and DyCrO3 (i.e. HoCr0.7Fe0.3O3 and DyCr0.7Fe0.3O3) powder samples were synthesized via a solution route. The structural properties of the samples were examined by Raman spectroscopy and X-ray diffraction techniques, which were further confirmed using the first-principle calculations. The dc magnetic measurements indicate that the Cr3+ ordering temperatures for the HoCrO3, HoCr0.7Fe0.3O3, and DyCr0.7Fe0.3O3 samples were 140 K, 174 K, and 160 K, respectively. The ac magnetic measurements not only confirmed the Cr3+ ordering transitions in these samples (obtained using dc magnetic measurements), but also clearly showed the Ho3+ ordering at ∼10 K in the present HoCrO3 and HoCr0.7Fe0.3O3 samples, which to our knowledge, is the first ac magnetic evidence of Ho3+ ordering in this system. The effective magnetic moments were determined to be 11.67μB, 11.30μB, and 11.27μB for the HoCrO3, HoCr0.7Fe0.3O3, and DyCr0.7Fe0.3O3 samples, respectively. For the first time, the magnetocaloric properties of HoCrO3 and HoCr0.7Fe0.3O3 were studied here, showing their potential for applications in magnetic refrigeration. In an applied dc magnetic field of 7 T, the maximum values of magnetic entropy change were determined to be 7.2 (at 20 K), 6.83 (at 20 K), and 13.08 J kg−1 K−1 (at 5 K) and the relative cooling power were 408, 387, and 500 J kg−1 for the HoCrO3, HoCr0.7Fe0.3O3, and DyCr0.7Fe0.3O3 samples, respectively.
 Please wait while we load your content...
Please wait while we load your content...

