
Development of an optimized HPLC method for the simultaneous determination of six compounds containing β-lactam ring in human plasma and urine using experimental design methodology†
Abstract
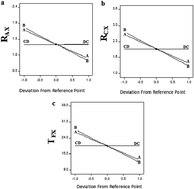
β-Lactam antibiotics are commonly prescribed with β-lactamase inhibitors to patients, for that it is necessary to develop an optimized chromatographic method which determine them simultaneously in biological fluids. In the present study a gradient HPLC method was demonstrated for the simultaneous separation and quantification of four commonly used β-lactam antibiotics and two β-lactamase inhibitors in human plasma and urine using statistical experimental design. A fractional factorial design was used in order to screen four independent factors: percentage of acetonitrile in the gradient program, pH of the aqueous phase, column temperature and percentage of trifluoroacetic acid (TFA) in aqueous phase. Examined factors were identified as significant using ANOVA analysis except column temperature and percentage of TFA in aqueous phase. The optimum condition of separation was determined with aid of central composite design. Chromatographic separation was achieved on Discovery® C18 column 5 μm (25 cm × 4.6 mm) with UV detection at 225 nm. In this method, the analytes were extracted from plasma and urine using solid phase extraction. The method was found to be linear, specific, precise and accurate. Tinidazole (TZ) was used as an internal standard (I.S.).
 Please wait while we load your content...
Please wait while we load your content...

