
Simultaneous preconcentration of uranium and thorium in aqueous samples using cloud point extraction
Abstract
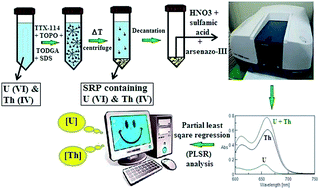
Uranium (U) and thorium (Th) are both chemically and radiologically toxic even at ultratrace concentrations. Hence, the development of new preconcentration procedures for their precise determination by simple, versatile and cost effective analytical techniques is desirable. A novel, simple and simultaneous cloud point extraction (CPE) procedure has been developed for preconcentrating trace amounts of U and Th in aqueous samples. Preconcentration of the metal ions in the surfactant rich phase of Triton X-114 was carried out by complexing them with trioctylphosphine oxide (TOPO) and N,N,N′,N′-tetraoctyldiglycolamide (TODGA). The preconcentrated solution was subjected to UV-visible spectrophotometry employing arsenazo-III. Partial least square regression analysis was then utilized to resolve their overlapping absorbance spectra and thereby allowing their determination in the presence of one another. The CPE procedure was optimized with respect to: pH of the solution, ionic strength, extraction temperature, phase separation temperature and concentrations of extractants, surfactant and co-surfactant. The developed CPE procedure resulted in percentage extraction efficiencies (EEs) of 98.0 ± 0.5 for U and 99.5 ± 0.5 for Th. Interference studies were also carried out and it was found that the recoveries of U and Th were 98% and 99% respectively in the absence of and ≥95% in the presence of interfering ions. The linear dynamic concentration ranges of the procedure were found to be 15–1000 ng mL−1 and 10–1000 ng mL−1 for U and Th, respectively. The developed methodology was successfully employed for the determination of U and Th in unspiked and spiked samples of ground water, lake water and sea water with ≤4% relative standard deviations. These samples were also directly analyzed by inductively coupled plasma mass spectrometry (ICP-MS) and the agreement between these two results at the 95% confidence level validates the developed methodology. The proposed CPE procedure can be used effectively for the simultaneous extraction of U and Th quantitatively with PFs of 94 for U and 100 for Th and can tolerate much higher levels of interfering ions.
 Please wait while we load your content...
Please wait while we load your content...

