
Modelling linear and branched amphiphilic star polymer electrolyte membranes and verification of the bond counting method
Abstract
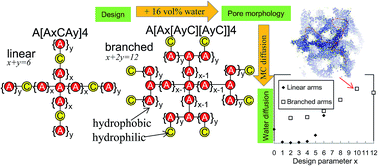
Polymer aggregation at a water content of 16% by volume is modelled by dissipative particle dynamics. The polymers contain a central hydrophobic bead from which four identical arms sprout out. For type L architectures each arm contains six hydrophobic A and one hydrophilic C bead whose position is varied ([A6−yCAy], 0 ≤ y ≤ 6). Each arm for type B architectures is two times larger and contains two branches with end grafted hydrophilic beads (A12−2y[AyC][AyC], 0 ≤ y < 6). Type L architectures reveal the narrowest pores when the C beads are located near the middle of each arm, while for type B they decrease with y. Also six architectures with pairwise unequal linear arms are considered. Among all 19 architectures the best connected hydrophilic clusters, derived from Monte Carlo tracer calculations through the pore networks, are obtained for type B architecture which contain short [AC] branches. The results are explained by calculating for each architecture the average number of A–A and A–C bonds that A beads are separated from the nearest C bead in each architecture. These insights might guide pore network design strategies in order to develop alternative polymer electrolyte membranes with low percolation thresholds for proton diffusion.
 Please wait while we load your content...
Please wait while we load your content...

