
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Immobilisation of water-oxidising amphiphilic ruthenium complexes on unmodified silica gel†
A.
Králík
,
M.
Hansen
and
B.
König
*
Institute of Organic Chemistry, University of Regensburg, D-93040 Regensburg, Germany. E-mail: burkhard.koenig@ur.de
First published on 7th January 2016
Abstract
Amphiphilic ruthenium complexes that catalyse the oxidation of water by light or cerium(IV) are non-covalently immobilised on untreated, commercial silica gel. The heterogeneous catalysts are prepared easily in one step and require no additives. High affinity of the amphiphilic complexes to silica gel leads to their rapid absorption from chloroform solution on silica gel. The complexes maintain immobilised in aqueous solution and their oxygen generation activity is comparable to water-soluble analogues. Within the tested range, the catalytic activity of the immobilised complexes does not depend on the surface loading. The simple technique facilitates the recovery and recycling of precious metals used in catalytic water oxidation.
Introduction
Global energy consumption is constantly increasing and renewable carbon-neutral alternatives to fossil fuels have to be found. Naturally, sunlight is the most promising renewable source of energy. It is available worldwide and its energy supply greatly exceeds the needs of our society.1,2 However, the major challenge is to develop an efficient method for storage of the harvested energy, so it can be utilised also during night and transported. One of the possible forms of storage are chemical fuels, compounds with high energy density. From the industrial point of view, water could be a cheap and abundant source of oxygen, as well as electrons, which can be used to produce e.g. hydrogen or methanol by a reduction of CO2. For research purposes, this process of water splitting can be divided into two parts that are studied separately, i.e. oxidative production of dioxygen and reductive production of dihydrogen.3,4Water oxidation is especially challenging, requiring four-electron transfers accompanied by a formation of highly-reactive oxygen species.5 Related heterogeneous photocatalytic systems use photoelectrochemical cells,6,7 photoactive semiconductors8,9 or metal-oxide catalysts coupled with metal-complex photosensitisers,10,11 while homogeneous systems usually consist of two main components – a photoredox-active photosensitiser (PS) and a water oxidation catalyst – together with a sacrificial electron acceptor, which is most often the peroxodisulfate anion.4 These can be represented by two separate water-soluble metal complexes.12 A proposed mechanism for this type of reaction can be seen in Fig. 1. This combination allows a rapid screening of different photosensitiser–catalyst couples. However, its activity is strongly dependent on concentration, since the required electron transfer between complexes is diffusion-controlled. To overcome this obstacle, several systems where the photosensitiser and the catalyst are covalently bound have been developed leading to a more efficient electron transfer.13–16 On the other hand, each combination of photosensitiser and catalyst requires a specific synthetic preparation, which makes screenings and complex variations notably difficult. We have recently developed self-assembled systems both for water oxidation and reduction based on amphiphilic metal complexes embedded in a phospholipid vesicular membrane, which increases the local concentration of photosensitiser and catalyst maintaining the activity even at low concentrations. The non-covalent interactions allow the facile variation of the system.17,18
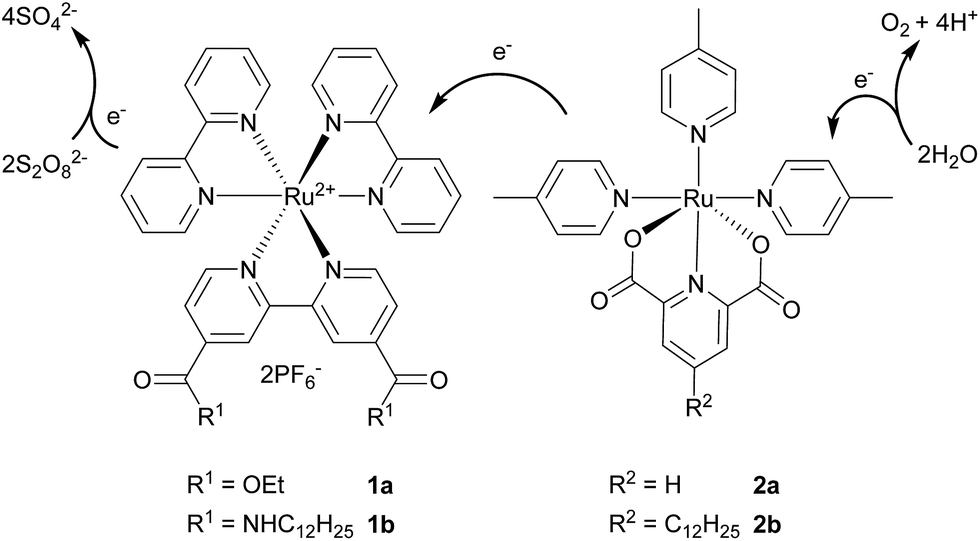 | ||
| Fig. 1 Structures of ruthenium photosensitiser (1) and catalyst (2), and a proposed mechanism for photocatalytic water oxidation. | ||
In recent years, immobilisation of metal complexes on the surface of inert materials, mostly silica gel, is of particular interest as a way of turning existing homogeneous catalytic systems into heterogeneous ones.19 This can be achieved covalently over Si–O bonds20 or non-covalently via hydrogen bonds.21,22 A different approach known as supported ionic liquid phase (SILP) uses catalytic metal complexes dissolved in a layer of ionic liquid immobilised on a solid supporting material.23,24 During our study focused on an application of SILP in photocatalytic water oxidation, a strong affinity of amphiphilic metal complexes to an untreated silica gel was observed. This phenomenon where amphiphilic surfactants bearing long alkyl chains are attracted to the polar surface of bare silica was already described in studies focused on HPLC separation.25–27 Gratifyingly, the immobilised complexes also maintain their catalytic activity. Therefore, we present a heterogeneous photocatalytic system for water oxidation based on amphiphilic ruthenium complexes as the photosensitiser (1b, Fig. 1) and the catalyst (2b) immobilised on a surface of silica gel. This setup overcomes several drawbacks. It can be prepared in one easy step, allows variations in complex types and ratios, and the heterogeneous nature may allow a facile recovery and recycling.
In a recently published study, a similar system utilising amphiphilic photosensitiser and cobalt catalyst non-covalently immobilised via hydrophobic interactions on a surface of silica gel modified with long alkyl chains in a presence of anionic or cationic surfactants was used for photocatalytic hydrogen evolution from water.28 However, our approach works with an unmodified silica gel and does not require additional additives.
Results and discussion
Immobilisation and stability
High affinity of the amphiphilic complexes 1b and 2b to silica gel can be already observed during the process of immobilisation. Both complexes are well soluble in chloroform forming a bright orange solution. After an addition of silica gel and a few minutes of stirring, significant decolourisation of the solution occurs, leaving the orange colour concentrated on the silica gel grains. The amount of adsorbed dye has been quantified using UV-Vis spectroscopy.During this experiment, different amounts of silica gel have been added to a 0.1 mM solution of photosensitiser 1b and after 5 minutes of stirring, the liquid phase has been analysed. Results are summarised in Table 1.
Using 50 mg of silica gel, corresponding to a loading of 10 μmol of photosensitiser per gram of silica gel, or more, results in almost quantitative adsorption leaving a colourless solution and providing orange-coloured solid particles.
Stability in aqueous solutions, in which the amphiphilic complexes are insoluble, was also investigated. Since a local lowering of pH can occur during the water oxidation reaction, neutral and acidic condition were chosen. Suspensions of silica gel with the immobilised complex 1b (20 μmol g−1) in aqueous buffers at pH 1, 4 and 7, respectively, were stirred for 48 hours. Table 2 shows the results confirming the high stability of the immobilised system in aqueous solutions. On the other hand, experiments conducted in acetonitrile and DMSO showed that in polar aprotic solvents, the complexes are washed off the silica gel surface.
A similar set of experiments in acetonitrile was conducted for water-soluble complexes 1a and 2a, which are insoluble in chloroform. No significant absorption from acetonitrile solution and a strong leakage into water was observed.
Performance in photocatalytic water oxidation
The immobilised metal complexes maintain their photocatalytic water oxidation activity, which is comparable to experiments in homogeneous solution both in terms of turnover numbers (TON) and reaction rate (Fig. 2). However, the fast photochemical degradation of the sensitiser molecule, limiting the photocatalysis to about 6 minutes, remains unchanged.17 Experiments using silica gel with different amounts of immobilised complexes showed that the activity of the system, in terms of oxygen evolution, is independent on the loading in a tested range from 20 to 2 μmol of 1b and 2 to 0.2 μmol of 2b per gram of support (in all cases, the photosensitiser to catalyst ratio is 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and also on the silica gel type.
:1) and also on the silica gel type.
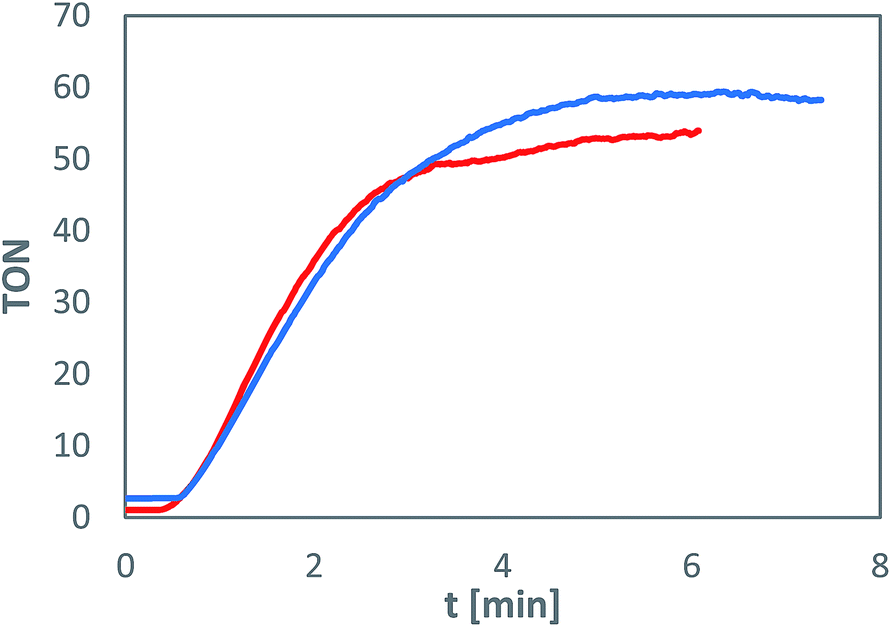 | ||
| Fig. 2 Comparison of oxygen evolution using a homogeneous system with water-soluble complexes 1a and 2a (red) and the heterogeneous system using silica-gel-bound complexes 1b and 2b (blue) monitored in situ by an oxygen probe. | ||
Results are summarised in Table 3. Loadings below this range showed very low oxygen production due to an insufficient light penetration of the sample, because of large amount of solids.
These observations also suggest that the silica gel surface is not evenly covered. In a case of an even coverage at the lowest functional loading, the calculated surface concentrations are 4 × 10−13 mol cm−2 for the photosensitiser and 4 × 10−14 mol cm−2 for the catalyst, respectively, implying a monolayer distribution with an average distance between the metal complexes around 20 nm. However, the required distance for an efficient electron transfer between a photosensitiser and a catalyst is much shorter.29 This suggests that the complexes are forming specific hot-spots with increased local concentration, which helps to maintain the functionality even at lower loadings.
All experiments have been repeated three times with standard deviations under 5% confirming the reproducibility of the results. The determined TON values based on the water oxidation catalyst lie between 30 and 50, which corresponds to previous homogeneous experiments utilising water-soluble complexes.17 The fluctuations are caused by different experimental setups for each loading with varying ratios of solid to liquid phase.
To prove that the system's short lifetime is caused by a degradation of the photosensitiser, a silica-gel-bound catalytic system bearing 20 μmol of photosensitiser and 2 μmol of the water oxidation catalyst per gram that has been previously used in a water oxidation reaction was filtered off, washed, dried and used to immobilise a batch of fresh photosensitiser.
This system was then fully functional for water oxidation reaching TON of 40, which corresponds to 84% of the original value.
Performance in chemical water oxidation
The use of this immobilisation method for chemical water oxidation reaction with ruthenium complex 3 (Fig. 3) as the catalyst and cerium ammonium nitrate as the oxidising agent was investigated. Since the complex is poorly soluble in water, mixtures containing acetonitrile or trifluoroethanol had to be used for published homogeneous experiments.30,31 The catalyst immobilised on silica gel is functional in solely aqueous solutions.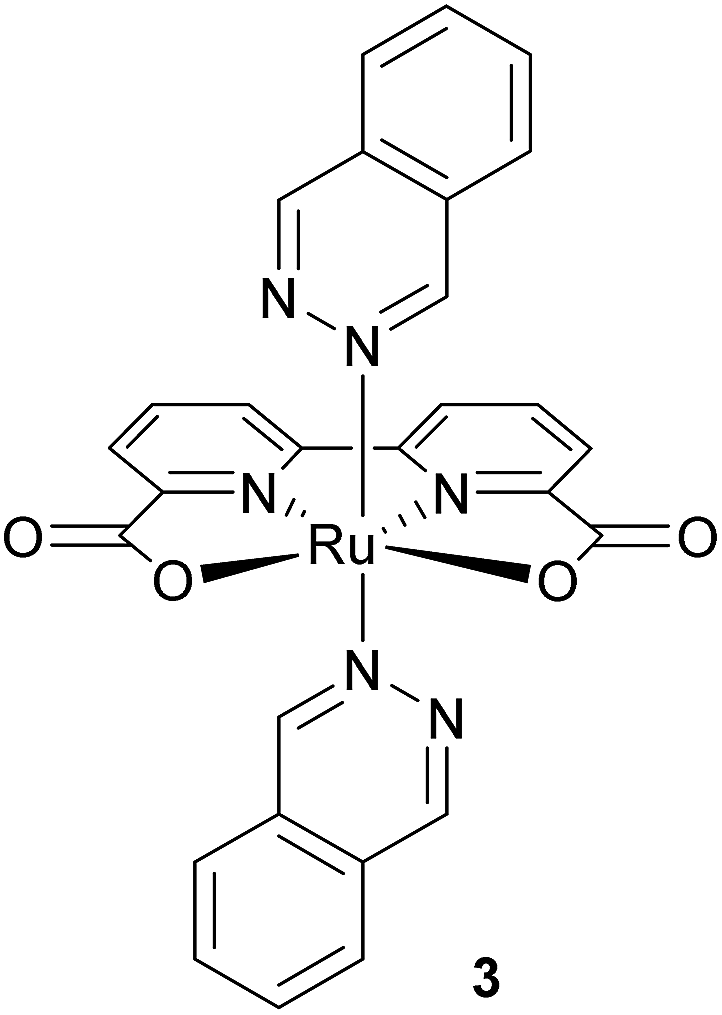 | ||
| Fig. 3 Structure of a ruthenium complex used for chemical water oxidation. | ||
Complex 3 shows the same affinity to silica gel as previously described for photosensitiser 1b – a complete decolourisation of its chloroform solution by an addition of the support was observed.
Table 4 shows that the activity of the system, in terms of oxygen production, is independent on the loading at a tested range from 2 to 0.1 μmol of catalyst per gram of support. All the experiments have been conducted three times with standard deviations mostly under 10% (20% for the loading of 0.1 μmol per gram), so they again show a good reproducibility. The obtained TON values are around 25000, which is lower compared with the homogeneous experiment where values of TON reached 40000 for our experiments and 55000 as published.31
| Loading of 3 [μmol g−1] | Amount of SiO2 [mg] | TON |
|---|---|---|
| Homogeneous | — | 41000 |
| 2 | 2 | 29000 |
| 1 | 4 | 24000 |
| 0.5 | 8 | 26000 |
| 0.2 | 20 | 17000 |
| 0.1 | 40 | 25000 |
Conclusions
A novel heterogeneous catalytic system for photocatalytic water oxidation is obtained by immobilising amphiphilic ruthenium complexes non-covalently on the surface of silica gel. The system is easy to prepare and the complexes maintain their catalytic activity, which is comparable to their water-soluble equivalents. However, the low stability of the photosensitiser under the reaction conditions still remains.Likewise, the immobilisation of a catalyst for chemical water oxidation is possible, showing good activity. This simple immobilisation technique may facilitate the recovery and regeneration of the catalysts.
Experimental
Preparation of silica-gel-based system for photocatalytic water oxidation
Stock solutions of photosensitiser 1b (2 mL, 1 mM) and catalyst 2b (200 μL, 1 mM), both in chloroform, were pipetted into a 10 mL round-bottom flask, diluted with chloroform to ca. 5 mL and silica gel (see Table 5 for amounts for different loadings) was added to the solution under stirring. This suspension was kept stirring for another 5 min, the solvent was then slowly evaporated under a mild vacuum (500 mbar at 40 °C) and finally dried in high vacuum (<0.5 mbar, 1 hour).| Loading of PS [μmol g−1] | Amount of SiO2 [mg] |
|---|---|
| 20 | 100 |
| 10 | 200 |
| 5 | 400 |
| 2 | 1000 |
| Component | Concentration |
|---|---|
| Photosensitiser (1) | 125 μM |
| Catalyst (2) | 12.5 μM |
| Na2S2O8 | 2.5 mM |
| Phosphate buffer | 50 mM |
Adsorption studies
Stock solution of photosensitiser 1b (500 μL, 1 mM) in chloroform was diluted to 5 mL, transferred into a 10 mL round-bottom flask and silica gel (25, 50 or 100 mg, pore size 60 Å, particle size 63–200 μm) was added to the solution under stirring. After five minutes, the suspension was filtered and UV-Vis spectra of the filtrate were measured. Concentration of the PS complex was determined by a comparison with a 5 μM standard solution and its peak absorbance at 287 nm.Stability studies
Silica gel (13 mg) bearing 20 μmol of photosensitiser 1b per gram was added to 2 mL of aqueous buffer (pH 1.0, 4.0 or 7.0) in a small vial with a stirrer. The vial was closed, covered in an aluminium foil and the suspension was kept stirring for 48 hours. Then, it was filtered and UV-Vis spectra of the filtrate were measured. Concentration of the PS complex was determined by a comparison with a 5 μM standard solution and its peak absorbance at 287 nm.The same process was used for stability measurements in DMSO and acetonitrile.
Photocatalytic water oxidation – homogeneous kinetic studies
Stock solutions of photosensitiser 1a (260 μL, 1 mM) and catalyst 2a (26 μL, 1 mM), both in chloroform, were pipetted into a small vial, evaporated on a shaker with a heating block and dried in high vacuum. Then, phosphate buffer (2.1 mL, 50 mM, pH 7.0) and sodium persulfate stock solution (5.25 μL, 1 M) were added (Table 6). The mixture was degassed by bubbling with nitrogen for ca. 10 minutes and it was transferred under a nitrogen atmosphere to a 2 mL vial equipped with a fluorescent spot for oxygen-probe measurements. The vial was filled to the top, so no air bubble was left inside to influence the results, closed, fastened under an upside down magnetic stirrer, its bottom was attached to a glass rod with a blue LED on the other side and the optical fiber of the oxygen detector was connected to the fluorescent spot. The reaction mixture was stirred and irradiated until the oxygen evolution ceased.Photocatalytic water oxidation – heterogeneous kinetic studies
The previously prepared silica-gel-based catalyst (13 mg) was weighed into a small vial. Then, phosphate buffer (2.1 mL, 50 mM, pH 7.0) and sodium persulfate stock solution (5.25 μL, 1 M) were added. The suspension was degassed by bubbling with nitrogen for ca. 10 minutes and it was transferred under nitrogen atmosphere to a 2 mL vial equipped with a fluorescent spot for oxygen-probe measurements, which was carried out in a same way as described in the previous paragraph.Photocatalytic water oxidation – heterogeneous quantitative studies
The previously prepared silica-gel-based catalyst (different amounts for different loadings, see Table 3) was weighed into a small crimp-top vial. Then, phosphate buffer (4.5 mL, 50 mM, pH 7.0) and sodium persulfate stock solution (11.3 μL, 1 M) were added. The vial was sealed and the suspension was degassed by bubbling with argon for ca. 15 minutes. Then, it was irradiated for 20 minutes using an array of six blue LEDs and cooled by a tap water in an aluminium block. The samples were left for an hour to equilibrate back to laboratory temperature and the produced oxygen was quantified using head-space GC.Reactivation
Collected suspensions containing silica gel with the highest loading of complexes used previously in water oxidation reaction were filtered, washed with distilled water until the filtrate was colourless and the solid residue was dried in high vacuum. Stock solution of photosensitiser 1b (1 mL, 1 mM) in chloroform was pipetted into a round 10 mL flask, diluted with chloroform to ca. 5 mL and the reused silica gel (50 mg) was added to the solution under stirring. This suspension was kept stirring for another 5 min, the solvent was then slowly evaporated under mild vacuum (500 mbar at 40 °C) and finally dried in high vacuum (<0.5 mbar, 1 hour).This treated silica gel (30 mg) was weighed into a small crimp-top vial and phosphate buffer (4.5 mL, 50 mM, pH 7.0) and sodium persulfate stock solution (11.3 μL, 1 M) were added. The vial was sealed; the suspension was degassed by bubbling with argon for ca. 15 minutes and then, an oxygen evolution reaction was carried out in the same way as described in the previous paragraph.
Preparation of silica-gel-based system for chemical water oxidation
Chloroform stock solution of catalyst 3 (10 μL, 1 mM) was pipetted into a 10 mL round-bottom flask, diluted with chloroform to ca. 5 mL and silica gel (see Table 7 for amounts for different loadings) was added to the solution under stirring. This suspension was kept stirring for another 5 min, the solvent was then slowly evaporated under mild vacuum (500 mbar at 40 °C) and finally dried in high vacuum (<0.5 mbar, 1 hour).| Loading of catalyst [μmol g−1] | Amount of SiO2 [mg] |
|---|---|
| 2.0 | 50 |
| 1.0 | 100 |
| 0.5 | 200 |
| 0.2 | 500 |
| 0.1 | 1000 |
Homogeneous chemical water oxidation
Degassed stock solution of catalyst 3 (4 μL, 1 mM) in trifluoroethanol was added via syringe into a small crimp-top vial previously purged with argon, then degassed stock solution of cerium ammonium nitrate in pH 1 triflic acid (2 mL, 500 mM) was added. The mixture was stirred for 1 hour and the produced oxygen was quantified using head-space GC.Heterogeneous chemical water oxidation
The previously prepared silica-gel-based catalyst (different amounts for different loadings, see Table 4) was weighed into a 20 mL crimp-top vial filled with argon, which was sealed and purged with argon again for 5 minutes. Then, a degassed stock solution of cerium ammonium nitrate in pH 1 triflic acid (2 mL, 500 mM) was added. The mixture was stirred for 1 hour and the produced oxygen was quantified using head-space GC.Acknowledgements
AK thanks the Fonds der Chemischen Industrie (FCI) for a stipend; we thank the Deutsche Forschungsgemeinschaft for financial support of the project.Notes and references
- J. Barber and P. D. Tran, J. R. Soc., Interface, 2013, 10, 20120984 CrossRef PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- P. D. Frischmann, K. Mahata and F. Wurthner, Chem. Soc. Rev., 2013, 42, 1847–1870 RSC.
- T. J. Meyer, Nature, 2008, 451, 778–779 CrossRef CAS PubMed.
- O. Khaselev and J. A. Turner, Science, 1998, 280, 425–427 CrossRef CAS PubMed.
- D. K. Zhong, J. Sun, H. Inumaru and D. R. Gamelin, J. Am. Chem. Soc., 2009, 131, 6086–6087 CrossRef CAS PubMed.
- S. U. M. Khan, M. Al-Shahry and W. B. Ingler, Science, 2002, 297, 2243–2245 CrossRef CAS PubMed.
- Z. Zou, J. Ye, K. Sayama and H. Arakawa, Nature, 2001, 414, 625–627 CrossRef CAS PubMed.
- K. Kalyanasundaram and M. Grätzel, Angew. Chem., Int. Ed., 1979, 18, 701–702 CrossRef.
- W. J. Youngblood, S.-H. A. Lee, Y. Kobayashi, E. A. Hernandez-Pagan, P. G. Hoertz, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, J. Am. Chem. Soc., 2009, 131, 926–927 CrossRef CAS PubMed.
- D. J. Wasylenko, R. D. Palmer and C. P. Berlinguette, Chem. Commun., 2013, 49, 218–227 RSC.
- F. Li, Y. Jiang, B. Zhang, F. Huang, Y. Gao and L. Sun, Angew. Chem., Int. Ed., 2012, 51, 2417–2420 CrossRef CAS PubMed.
- N. Kaveevivitchai, R. Chitta, R. Zong, M. El Ojaimi and R. P. Thummel, J. Am. Chem. Soc., 2012, 134, 10721–10724 CrossRef CAS PubMed.
- M. R. Norris, J. J. Concepcion, D. P. Harrison, R. A. Binstead, D. L. Ashford, Z. Fang, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2013, 135, 2080–2083 CrossRef CAS PubMed.
- D. L. Ashford, D. J. Stewart, C. R. Glasson, R. A. Binstead, D. P. Harrison, M. R. Norris, J. J. Concepcion, Z. Fang, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2012, 51, 6428–6430 CrossRef CAS PubMed.
- M. Hansen, F. Li, L. Sun and B. Konig, Chem. Sci., 2014, 5, 2683–2687 RSC.
- S. Troppmann and B. König, Chem.–Eur. J., 2014, 20, 14570–14574 CrossRef CAS PubMed.
- A. Kirschning, Immobilized Catalysts, Springer, Germany, 2004 Search PubMed.
- A. Jana, J. Mondal, P. Borah, S. Mondal, A. Bhaumik and Y. Zhao, Chem. Commun., 2015, 51, 10746–10749 RSC.
- C. Bianchini, D. G. Burnaby, J. Evans, P. Frediani, A. Meli, W. Oberhauser, R. Psaro, L. Sordelli and F. Vizza, J. Am. Chem. Soc., 1999, 121, 5961–5971 CrossRef CAS.
- C. Bianchini, P. Barbaro, V. Dal Santo, R. Gobetto, A. Meli, W. Oberhauser, R. Psaro and F. Vizza, Adv. Synth. Catal., 2001, 343, 41–45 CrossRef CAS.
- C. P. Mehnert, R. A. Cook, N. C. Dispenziere and M. Afeworki, J. Am. Chem. Soc., 2002, 124, 12932–12933 CrossRef CAS PubMed.
- A. Riisagera, R. Fehrmanna, M. Haumannb and P. Wasserscheidb, Top. Catal., 2006, 40, 91–102 CrossRef.
- J. H. Knox and G. R. Laird, J. Chromatogr. A, 1976, 122, 17–34 CrossRef CAS.
- Y. Ghaemi and R. A. Wall, J. Chromatogr. A, 1979, 174, 51–59 CrossRef CAS.
- B. Juskowiak, J. Chromatogr. A, 1994, 668, 313–321 CrossRef CAS.
- C. Bachmann, B. Probst, M. Oberholzer, T. Fox and R. Alberto, Chem. Sci., 2015, 7, 436–445 RSC.
- B. König, Chemical Photocatalysis, De Gruyter, Berlin, 2013 Search PubMed.
- L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS PubMed.
- L. Duan, C. M. Araujo, M. S. G. Ahlquist and L. Sun, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15584–15588 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra24088c |
| This journal is © The Royal Society of Chemistry 2016 |