
Syntheses, structures and properties of five entangled coordination polymers constructed with trigonal N-donor ligands†
Yu Chen,
Juan Yang,
Xi-Chi Wang,
Yu-Ci Xu,
Hui-Ling Xu,
Xiao-Shan Zeng and
Dong-Rong Xiao*
College of Chemistry and Chemical Engineering, Southwest University, Chongqing, 400715, P. R. China. E-mail: xiaodr98@swu.edu.cn; Fax: +86-23-68254000
First published on 7th January 2016
Abstract
Based on the mixed-ligand system, five new entangled coordination polymers have been synthesized, namely, [Co1.5(1,3-bdc)1.5(tib)(H2O)] (1), [Ni(1,3-bdc)(tib)(H2O)2]·0.5H2O (2), [Cu(Hbtc)(Htpim)] (3), [Co(bpndc)(pytpy)(H2O)]·0.5H2O (4), and [Ni(bpndc)(pytpy)(H2O)]·0.5H2O (5), (1,3-bdc = 1,3-benzenedicarboxylate, btc = 1,3,5-benzenetricarboxylate, bpndc = 4,4′-benzophenone dicarboxylate, tib = 1,3,5-tris(1-imidazolyl)benzene, Htpim = 2,4,5-tri(4-pyridyl)-imidazole, pytpy = 2,4,6-tris(4-pyridyl)pyridine). Their structures were determined by single-crystal X-ray diffraction analyses and further characterized by elemental analyses, IR spectroscopy and TG analyses. Compound 1 exhibits a new trinodal (3,4,4)-connected 3D self-penetrating framework with (83)2(63.82.9)2(84.9.10) topology. Compound 2 displays an interesting 2D → 3D polythreading framework constructed from sidearm-containing 2D 63-hcb nets. Compound 3 exhibits an unusual (2D → 3D) entangled array with the coexistence of polythreading and interdigitation self-assembled from sidearm-containing 2D double-edged nets. Compounds 4 and 5 both show an intriguing 3-fold interpenetrated PtS-type (4,4)-connected 3D framework containing meso-helices. In addition, the magnetic properties of compounds 1–5 have been investigated in the temperature range 2–300 K.
Introduction
In recent years, the rational design and synthesis of metal–organic frameworks (MOFs) have been rapidly developed, in view of their extensive potential application in gas adsorption and separation,1 molecular magnetism,2 heterogeneous catalysis3 and nonlinear optics (NLO).4 Heretofore, a considerable amount of literature indicated that diverse structures and intriguing topologies presumably contribute to functional applications.3b,5 Thereupon a huge endeavour has been made to construct and develop different structures. Entangled networks,6 as one of the most active subclasses among MOFs, are flourishing and a vast number of entangled networks, such as interpenetration,7 self-penetrating8 and some new entanglements named polythreading,9 interdigitation,10 etc. have been intensively explored. Likewise, exploitation of entangled structures could be helpful for both modulating structures of MOFs and understanding the relationships between the entangled structure and function of MOFs.11To attain entangled coordination networks, it is crucial to take the factors, such as nature of metal, organic ligands, solvent system, pH value etc., into consideration.12 Among various organic ligands, the N-donor ligands as good candidates for fabricating entangled networks, have been introduced to synthetic strategy many times.13 To date, a variety of linear N-donor ligands, such as 4,4′-bipyridyl,14 1,2-bis(4-pyridyl)ethane,15 1,4-bis(imidazole-1-yl)butane,16 1,4-bis(1,2,4-triazol-1-yl)butane17 and 1,4-bis(imidazole-1-ylmethyl)benzene,8b have been widely used to construct numerous entangled networks. In contrast with linear ligands, trigonal N-donor ligands are relatively rarely employed to build entangled structures, and some literatures also reveal that MOFs constructed from trigonal N-donor ligands have no entanglements.18 Nevertheless, there are some successful entangled examples with trigonal N-donor ligands, for example, Mukherjee and co-workers have synthesized a 2-fold interpenetrated 2D network with tib (tib = 1,3,5-tris(1-imidazolyl)benzene, Scheme 1a);19 our group has reported one compound consisting of two identical 3D self-threading frameworks with CdSO4 topology based on Htpim (Htpim = 2,4,5-tri(4-pyridyl)-imidazole, Scheme 1b).20 Recently, Wang et al. have obtained a crystal with 2-fold interpenetrated three-dimensional framework via using pytpy (pytpy = 2,4,6-tris(4-pyridyl)pyridine, Scheme 1c).21 Therefore, in some degree, these trigonal N-donor ligands could be feasible to fabricate novel entangled structures for the followed merits: (i) On the one hand, used as large tritopic ligands, trigonal N-donor ligands could generate large voids that may result in entangled structures. (ii) On the other hand, they can act as long bidentate bridging ligands with side arms, which provide great possibilities for the formation of polythreading or interdigitation.
 | ||
| Scheme 1 Schematic drawing of tib, Htpim, pytpy, 1,3-H2bdc, H3btc and H2bpndc ligands. | ||
With the above stated aspects in mind, we mixed these three trigonal N-containing ligands (tib, Htpim, and pytpy) with aromatic polycarboxylate ligands to react with transition metal salts under hydrothermal conditions. By unceasing efforts of us, five novel entangled structures have been successfully synthesized, namely [Co1.5(1,3-bdc)1.5(tib)(H2O)] (1), [Ni(1,3-bdc)(tib)(H2O)2]·0.5H2O (2), [Cu(Hbtc)(Htpim)] (3), [Co(bpndc)(pytpy)(H2O)]·0.5H2O (4) and [Co(bpndc)(pytpy)(H2O)]·0.5H2O (5) (1,3-bdc = 1,3-benzenedicarboxylate, btc = 1,3,5-benzenetricarboxylate, bpndc = 4,4′-benzophenone dicarboxylate, Scheme 1). The syntheses, crystal structures and properties of these compounds and the effects of the N-donor ligands on the structures will be represented and discussed in this paper.
Experimental
Materials and methods
The chemicals and solvents were commercially purchased and used as received without further purification. The tib,22 Htpim23 and pytpy24 ligands were synthesized according to the reported procedures, respectively.Elemental analyses (C, H and N) were performed on a Perkin-Elmer 2400 CHN Elemental Analyzer. Ni, Co and Cu were determined by a tps-7000 Plasma-Spec(I) inductively coupled plasma-atomic emission spectrometer (ICP-AES). IR spectra were recorded in the range 400–4000 cm−1 on a Bio-Rad FTS-185 FT/IR spectrophotometer using KBr pellets. TG analyses were performed on a NETZSCH STA 449C instrument in flowing N2 with a heating rate of 10 °C min−1. XRPD data were recorded on a XD-3 diffractometer using Cu Kα radiation. Variable-temperature magnetic susceptibility data were obtained on a SQUID magnetometer (Quantum Design, MPMS-7) in the temperature range of 2–300 K with an applied field of 1.0 kOe. All the magnetic susceptibility data were corrected from diamagnetic contributions estimated from Pascal's constants.
Syntheses of compounds 1–5
X-Ray crystallography
Suitable single crystals of compounds 1, 2 and 5 were selected for single-crystal X-ray diffraction analysis (diffractometer device type: SuperNova, Dual, Cu at zero, EosS2). They were collected with Cu Kα radiation (λ = 1.54184 Å) at 290 K, 279 K and 292 K, respectively. Suitable single crystals of 3 and 4 were selected for single-crystal X-ray diffraction analysis (diffractometer device type: Bruker Smart Apex CCD). They were collected with Mo Kα radiation (λ = 0.71073 Å) at 293 K. Using Olex2,25 the structures of 2–5 were solved with the Superflip26 structure solution program using Charge Flipping, while the structure of 1 was solved with the ShelXS27 structure solution program using Direct Methods. Moreover, the structures of 1–5 were refined with the ShelXL27 refinement package using least square minimization. All non-hydrogen atoms were refined anisotropically. The organic hydrogen atoms were generated geometrically. The aqua hydrogen atoms were located from difference Fourier maps and refined with isotropic displacement parameters. The detailed crystallographic data and structure refinement parameters for compounds 1–5 are summarized in Table 1. Selected bond lengths for 1–5 are listed in Table S1.† The topological analyses were done with the TOPOS program.28| a R1 = ∑||F0| − |FC||/∑|F0|.b wR2 = ∑[w(F02 − FC2)2]/∑[w(F02)2]1/2. | |||||
|---|---|---|---|---|---|
| Compound | 1 | 2 | 3 | 4 | 5 |
| Empirical formula | C27H20Co1.5N6O7 | C23H21NiN6O6.5 | C27H17CuN5O6 | C35H26CoN4O6.5 | C35H26NiN4O6.5 |
| Mr (g mol−1) | 628.88 | 544.17 | 570.99 | 665.53 | 665.31 |
| Crystal system | Monoclinic | Orthorhombic | Monoclinic | Monoclinic | Monoclinic |
| Space group | C2/c | Pbca | C2/c | C2/c | C2/c |
| a (Å) | 28.3457(5) | 15.65677(16) | 23.2430(12) | 31.982(2) | 31.9008(6) |
| b (Å) | 11.56051(10) | 15.8377(2) | 9.9760(5) | 8.0580(6) | 7.98993(8) |
| c (Å) | 20.1709(4) | 17.8327(2) | 20.9258(11) | 28.638(2) | 29.2439(5) |
| α (°) | 90 | 90 | 90 | 90 | 90 |
| β (°) | 130.063(3) | 90 | 106.8580(10) | 122.4490(10) | 124.349(3) |
| γ (°) | 90 | 90 | 90 | 90 | 90 |
| V (Å3) | 5058.8(2) | 4421.92(9) | 4643.6(4) | 6228.0(7) | 6154.0(2) |
| Z | 8 | 8 | 8 | 8 | 8 |
| ρcalcd (g cm−3) | 1.651 | 1.635 | 1.633 | 1.420 | 1.436 |
| μ (mm−1) | 8.335 | 1.783 | 0.998 | 0.606 | 1.377 |
| F(000) | 2564.0 | 2248.0 | 2328 | 2744.0 | 2752.0 |
| 2Θ range per ° | 8.152–143.102 | 9.364–143.114 | 3.658–56.314 | 3.014–57.804 | 11.098–142.994 |
| Reflections collected | 46![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 039 039 |
38853 |
14227 |
18836 |
43596 |
| Unique data (Rint) | 4932(0.0484) | 4293(0.0398) | 5539(0.0209) | 7523(0.0272) | 5967(0.0327) |
| GOF on F2 | 1.056 | 1.079 | 1.026 | 1.046 | 1.052 |
| R1a/wR2b[I > 2σ (I)] | 0.0340/0.0847 | 0.0386/0.1029 | 0.0305/0.0788 | 0.0420/0.1160 | 0.0432/0.1312 |
| R1a/wR2b(all data) | 0.0388/0.0879 | 0.0395/0.1037 | 0.0407/0.0841 | 0.0658/0.1297 | 0.0446/0.1327 |
Results and discussion
Crystal structures
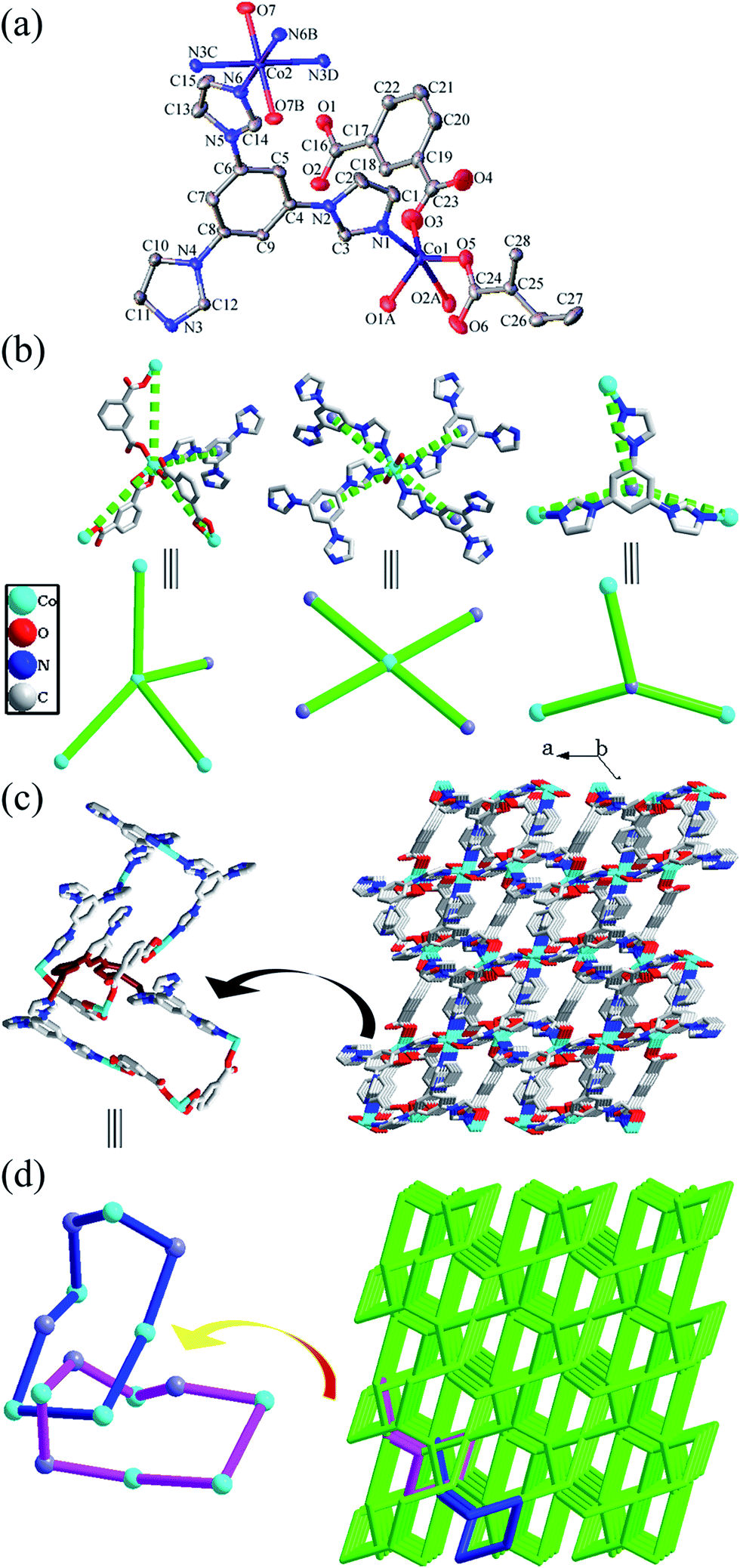 | ||
| Fig. 1 For compound 1: (a) ORTEP diagram showing the coordination environment for Co atoms in 1. (b) Perspective (upper) and simplified (down) views of the two different four-connected Co atoms and three-connected tib ligand. Perspective (c) and simplified (d) views of the (3,4,4)-connected 3D self-penetrating network with (83)2(63.82.9)2(84.9.10) topology. | ||
These different connection modes among Co atoms and ligands result in an interesting complicated 3D network (Fig. 1c). As depicted in Fig. 1b, both Co1 and Co2 centers can be considered as 4-connected nodes, and the tib ligands can be regarded as 3-connected nodes. So the resulting structure of 1 can be rationalized as a (3,4,4)-connected network with (83)2(63.82.9)2(84.9.10) topology, which represents a new topological prototype (Fig. 1d). Additionally, self-catenation of the 8-membered shortest rings by other 8-membered rings in self-penetrating network 1 is depicted in Fig. 1c and d.
There are intermolecular hydrogen bonding interactions between the coordinated water molecules and the carboxylate oxygen atoms (O7–H7A⋯O5 and O7–H7B⋯O4, Table S2†), which further stabilize the whole structure of 1.
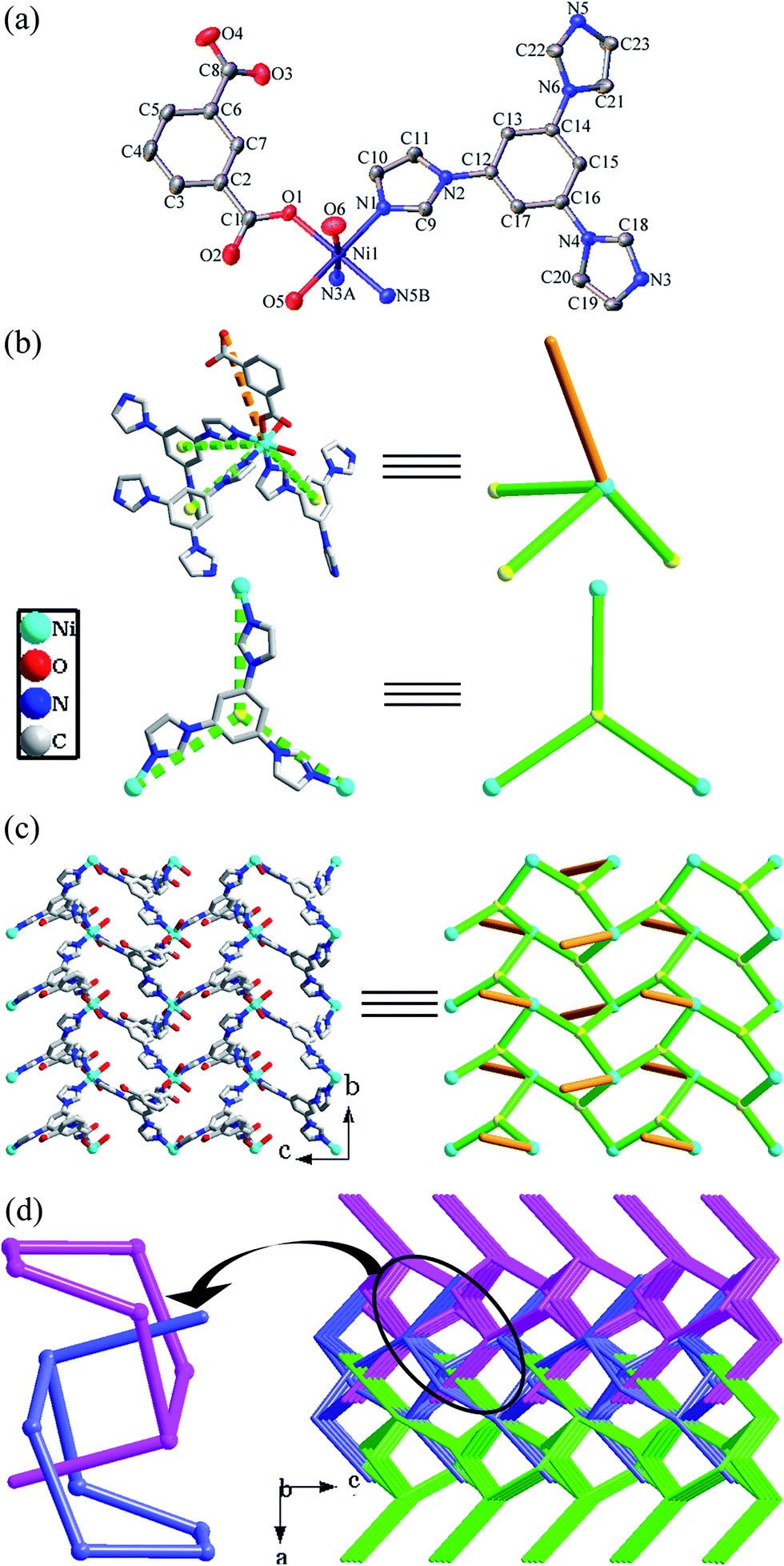 | ||
| Fig. 2 For compound 2: (a) ORTEP diagram showing the coordination environment for Ni atoms in 2. (b) Perspective (left) and simplified (right) views of the four-connected Ni atom and three-connected tib ligand. (c) Perspective (left) and simplified (right) views of a 2D network with distinct dangling arms (the arms of 1,3-bdc ligands are highlighted by orange lines). (d) The schematic representation of the (2D → 3D) polythreading network. | ||
The most outstanding feature for 2 is that the structure displays a 2D → 3D polythreading array assembled from 2D 63-hcb motifs. As shown in Fig. 2d, the 2D layers are stacked in a parallel ABAB sequence at a distance of 7.83 Å; while the lateral 1,3-bdc ligand has an effective length of 8.22 Å (Fig. S2a†). As a result, the dangling arms of each layer are threaded into the hexagonal cavities of two adjacent layers above and below. Every hexagonal void of each layer is just pierced by one arm coming from above or below layers alternately (Fig. S2c†). Finally, this unique mutual threading of adjacent layers in 2 leads to a novel 2D → 3D polythreading framework, originating from infinite sidearm-containing 2D 63-hcb networks (Fig. 2d). So far, such 2D → 3D polythreading frameworks are still very rare in the entangled systems. To the best of our knowledge, compound 2 represents the fourth 2D → 3D polythreading framework that is built from 2D 63-hcb networks.29
In addition, the water molecules and the carboxylate oxygen atoms participate in hydrogen bonding interactions to form (O6–H6B⋯O4, O7–H7A⋯O4 and O7–H7B⋯O4) hydrogen bonds (Table S2†).
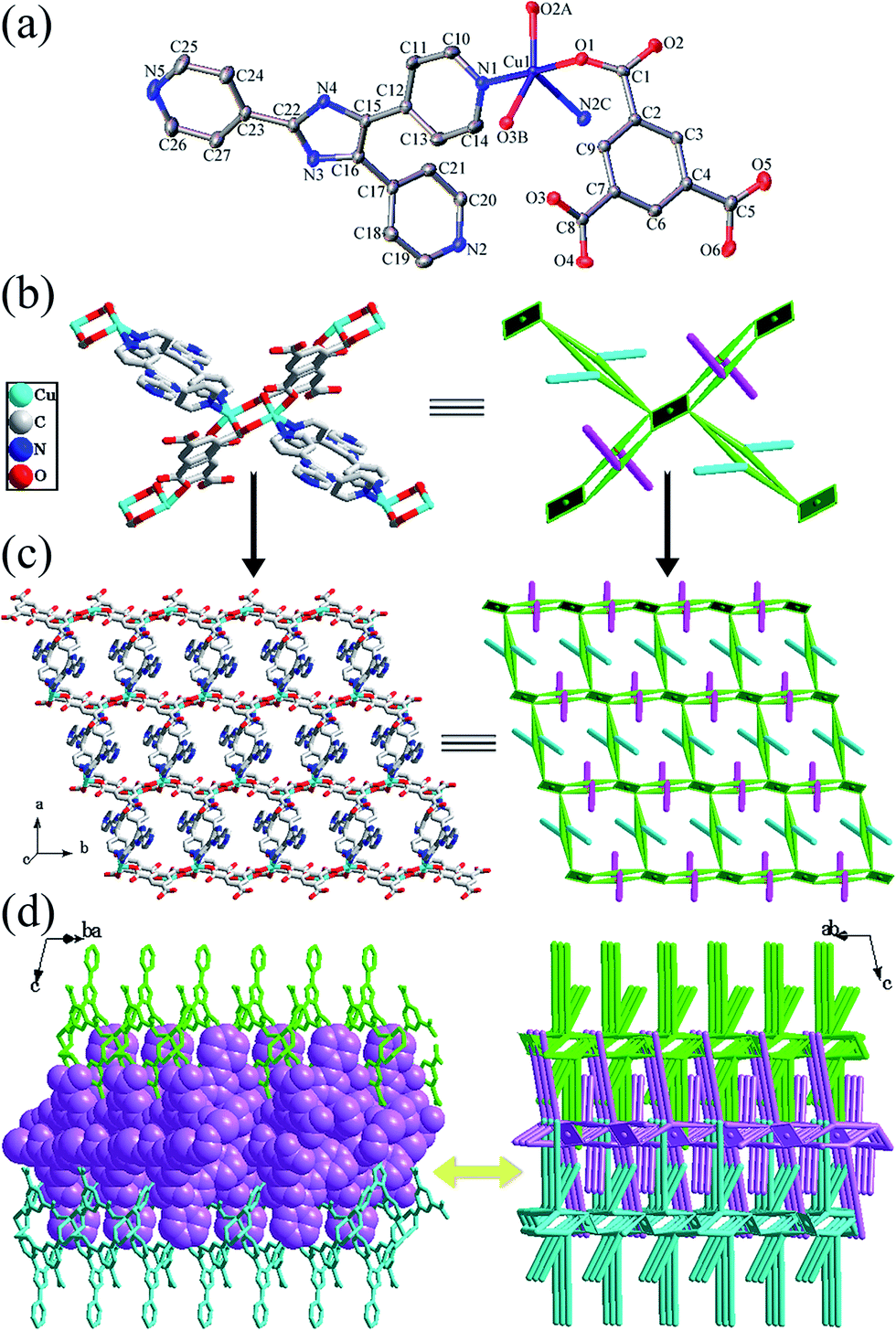 | ||
| Fig. 3 For compound 3: (a) ORTEP diagram showing the coordination environment for Cu atoms in 3. (b) Perspective (left) and simplified (right) views of the linkages of a dimetallic SBU with four equivalent neighbors. Four “double-bridges” serve as four double-edged linkers. (c) Perspective (left) and schematic views (right) of a single double-edged 2D network with two distinct dangling arms (the arms of Htpim ligands are highlighted by cerulean lines, the arms of Hbtc ligands are highlighted by purple lines). (d) Perspective and space-filling (highlighted by purple) (left) and schematic views (right) of the polythreading of three 2D layers. | ||
On the basis of these connection modes, two Cu(II) ions are connected by two μ2-carboxylate ends to form a dinuclear copper unit as a secondary building unit (SBU). The nonbonding Cu⋯Cu distance in the dinuclear copper unit is 4.444 Å. Each dimetallic SBU is further linked to four equivalent neighbors through two pairs of Hbtc ligands and two pairs of Htpim ligands (Fig. 3b), which orient in four different directions and each pair of them acts as “double-bridges” chelating to another SBU, to generate an unusual 2D network (Fig. 3c). Topologically each dimetallic SBU acts as a 4-connected node, and four “double-bridges” serve as four double-edged linkers (Fig. 3b). Then, this 2D network is finally defined as a 2D double-edged (4,4) network with two distinct lateral arms (Fig. 3c). The two distinct lateral arms have the effective lengths of ca. 5.82 Å and 10.36 Å respectively (Fig. S3a†). Moreover, the large tetragonal windows exist in the single double-edged network with maximum dimensions of 9.71 Å × 9.98 Å, which is formed by four dimetallic SBUs linked by two pairs of Hbtc ligands and two pairs of Htpim ligands as “double-bridges”. This remarkable feature of the single network is a necessary condition for the formation of polythreading and interdigitation. As shown in Fig. 3d, the 2D layers are stacked together in an parallel ABAB sequence with a interlayer distance of 10.01 Å. Interestingly, each tetragonal window of each layer is threaded by two long arms (i.e. the pyridine groups of Htpim ligands) and interdigitated by two short arms (i.e. the protonated carboxylate groups of Hbtc ligands) that belong to the two nearest adjacent layers (one above and one below) (Fig. 3d and S3b†). Then a new (2D → 3D) entangled network containing both polythreading and interdigitation characters is obtained, originating from infinite sidearm-containing 2D double-edged frameworks. To the best of our knowledge, a 2D double-edged layer as a structural motif is quite rare in entangled systems.30
Additionally, there are two kinds of hydrogen bonds in the structure (N4–H4⋯O4 and O6–H6⋯N5, Table S2†), which further stabilize the whole structure of 3.
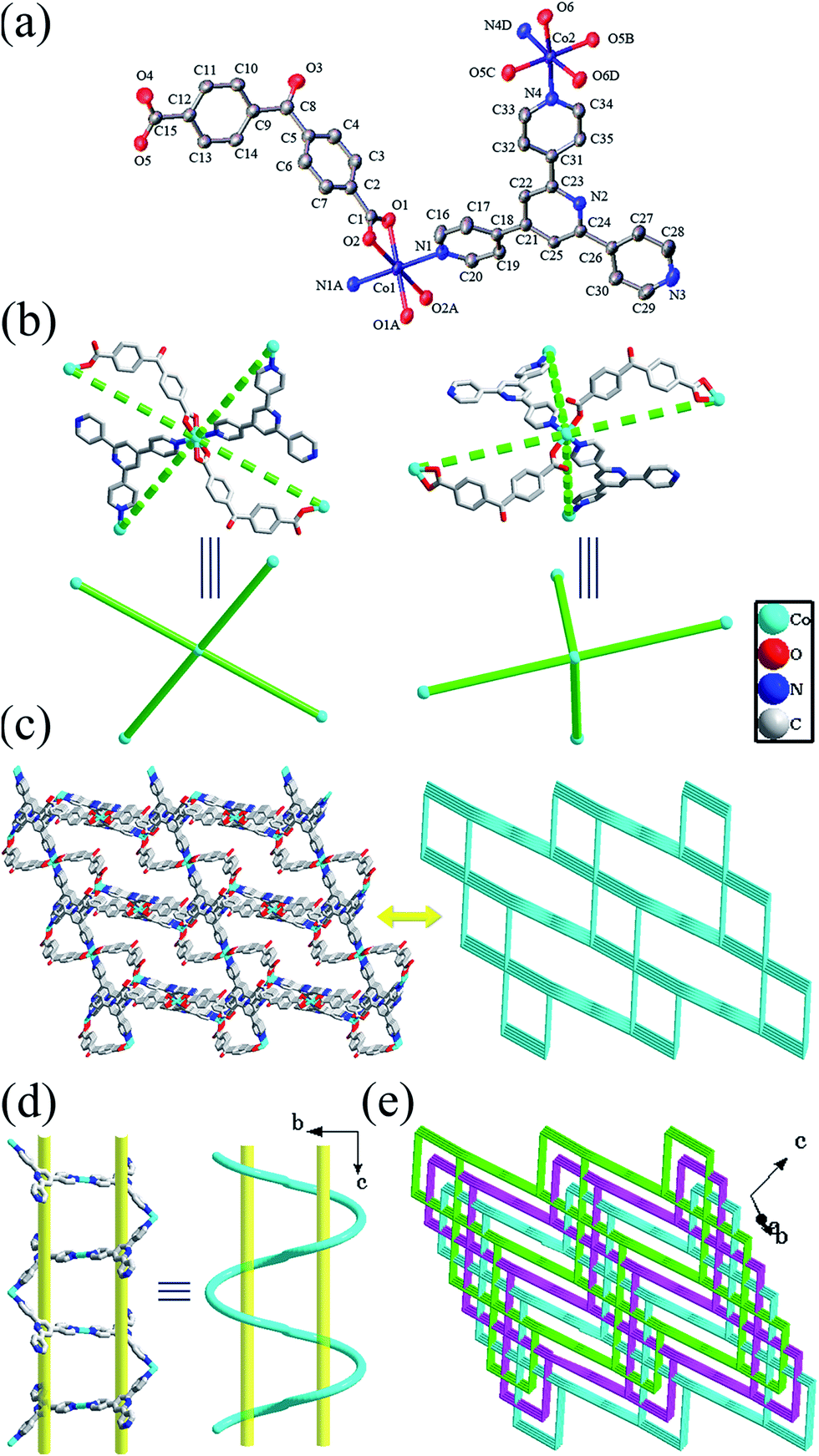 | ||
| Fig. 4 For compound 4: (a) ORTEP diagram showing the coordination environment for Co atoms in 4. (b) Perspective (upper) and simplified (down) views of the two different four-connected Co atoms. (c) Perspective (left) and simplified (right) views of a single 3D framework. (d) Perspective (left) and schematic (right) views of the meso-helix. (e) Scheme of the complete structure consisting of three interpenetrated nets with the pts topology. | ||
Topologically, the Co atoms can be regarded as four-connected nodes (Fig. 4b). So the resulting structure can be reduced to a (4,4)-connected net with the pts topology (Fig. 4c). To date, meso-helical structures are quite uncommon in pts net, and only one example has been reported by Yuan and Yang.31 Moreover, in order to minimize the large voids from a single 3D framework and stabilize the structure, three identical frameworks interpenetrate each other, thus giving a new 3-fold interpenetrated pts net, as depicted in Fig. 4e and S6.† An analysis of the interpenetration topology with the TOPOS program reveals that it belongs to Class Ia, that is, the three identical interpenetrated nets are generated only by translation and the translating vector is [010] (8.06 Å). Due to such threefold interpenetration, the solvent-accessible voids of 4 are radically reduced to be 8.1% of the crystal volume as calculated by PLATON.32
There are versatile hydrogen bonding interactions between the coordination water and pyridine nitrogen atoms, the lattice water and the carboxylate oxygen atoms, and the coordination water and the solvent water (Table S2†).
Effects of trigonal N-donor ligands
In this work, three trigonal N-containing ligands (tib, Htpim and pytpy) play a key role in determining the structures of the coordination polymers. In 1 and 2, three imidazole groups of the tib ligand participate in the coordination connecting three metal atoms to form 2D layers (Fig. S1 and S2b†). In 3, two of three pyridyl moieties of Htpim ligand link metal centers and third pyridyl moiety dangles as a lateral arm which can thread through a cavity from an adjacent layer (Fig. 3b). This means that trigonal ligands may provide the lateral arms to form the ploythreading structure, when the terminals of the ligands are not fully employed. In 4 and 5, the pytpy ligands also perform as V-shaped ditopic linkers; only two-thirds of the pyridine groups are used to bridge metal centers and generate a single-stranded meso-helical chain (Fig. S5†). Another example, reported by Ma and co-workers,33 confirms that trigonal N-donor ligands can be utilized to build meso-helical chains when they serve as bidentate bridging ligands. To sum up, these large trigonal N-donor ligands not only can generate large voids that may result in entangled structures, but also can act as long bidentate bridging ligands with side arms to provide great possibilities for the formation of polythreading or interdigitation.X-Ray powder diffractions and thermal properties
In order to check the phase purity of compounds 1–5, the X-ray powder diffraction (XRPD) patterns were recorded at room temperature. As shown in Fig. S7–S11,† the peak positions of simulated and experimental patterns are in good agreement with each other, demonstrating the phase purity of the products. The differences in intensity may be due to the preferred orientation of the crystalline powder samples.Thermal gravimetric analyses (TG) were performed on compounds 1–5 to investigate their thermal stabilities (Fig. S12–S16†). The TG curve of 1 exhibits three steps of weight losses (Fig. S12†). The first weight loss is 3.0% in the temperature range of 55–390 °C which corresponds to the loss of coordinated water molecules (calcd 2.86%). The second weight loss starts from 391 °C to 488 °C and the third weight loss is going on and continues up to 784 °C, both corresponding to the release of organic components. The total weight loss of 78.6% is less than the calculated value of 82.11% if the final product is assumed to be CoO, which indicates that the decomposing process is not complete due to the use of nitrogen protection. In the DSC curve of 1, the endothermic peak at 197 °C and the exothermic peak at 445 °C correspond to the loss of coordinated water molecules; the endothermic peak at 465 °C and the exothermic peaks at 525 °C and 705 °C are all related to the release of the organic ligands.
The TG curve of 2 is shown in Fig. S13,† the TG curve exhibits two steps of weight losses. The first weight loss is 8.57% from room temperature to 348 °C, which corresponds to the loss of the coordinated and non-coordinated water molecules (calcd 8.28%). Then the structure decomposes in the range of 349–670 °C to form NiO as the residue (observed 14.03%, calcd 13.73%). From the DSC curve of 2, we can sense that the endothermic peak at 189 °C corresponds to the loss of lattice water and coordinated water molecules; the exothermic peaks at 483 °C and 684 °C are both related to the release of the organic ligands.
The TG curve of 3 displays three steps of weight losses, as depicted in Fig. S14.† The first weight loss is 39.13% from the room temperature to 400 °C, then the second weight loss (25.0%) is going on and continues up to 524 °C, the last weight loss is going up to 784 °C with a weight loss of 19.94%, corresponding to the release of Hbtc ligands and Htpim ligands. The total weight loss of 84.07% is slightly less than the calculated value of 85.99% if the final product is assumed to be CuO, which indicate that the decomposing process is not complete due to the use of nitrogen protection. From the corresponding DSC curve of 3, the endothermic peak at 393 °C and exothermic peaks at 494 °C and 610 °C are all related to the release of Hbtc ligands and Htpim ligands.
The TG curves of 4 and 5 illustrate highly similar features in Fig. S15 and S16.† Both of these two curves exhibit two-step weight losses. For compound 4, the first step occurs from 41 °C until 372 °C, resulting in a 3.93% weight loss, while for compound 5, the mechanism starts at 39 °C until 389 °C, leading to a weight loss of 4.17%. These weight losses correspond to the loss of the coordinated and non-coordinated water molecules (both calcd 4.06%). The second weight losses start from 373 °C for 4 and 391 °C for 5, both corresponding to the release of organic components. For compounds 4 and 5, the total weight losses of 83.90% and 74.36% are less than the calculated value of 88.74% and 88.77% if the final products are assumed to be CoO and NiO, respectively, which indicates that the decomposing processes are not complete due to the use of nitrogen protection. In their DSC curves (Fig. S15 and S16†), the endothermic peak at 393 °C and the exothermic peaks at 504 °C, 623 °C, 693 °C for 4, the endothermic peak at 420 °C and the exothermic peaks at 483 °C, 571 °C, 631 °C, 691 °C for 5, are all related to the decomposition of the organic ligands.
Magnetic properties
The magnetic properties of compounds 1–5 were measured in the temperature range of 2–300 K at a direct current field of 1.0 kOe (Fig. 5 and Table 2).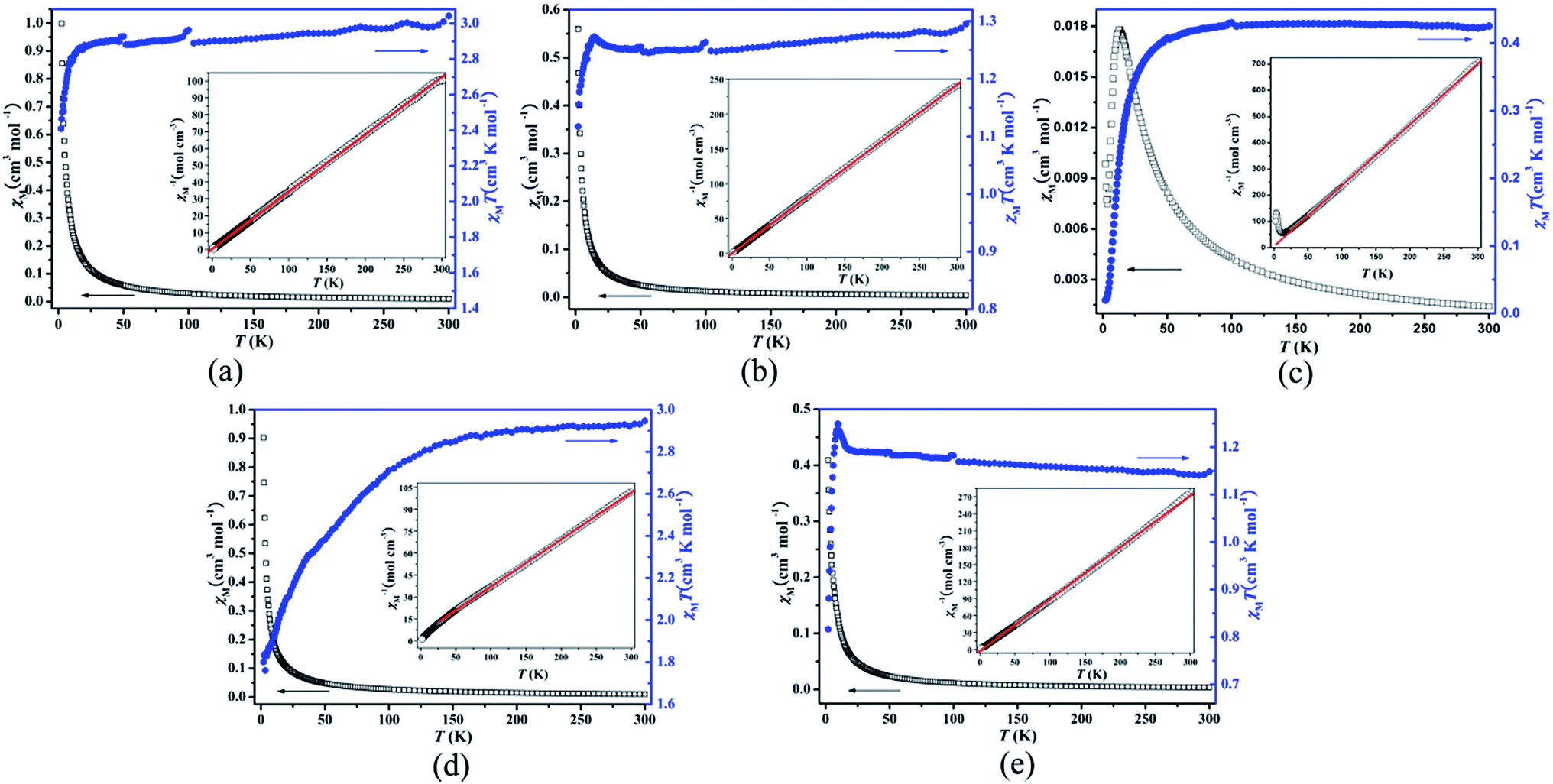 | ||
| Fig. 5 Thermal variation of χM and χMT for compounds 1 (a), 2 (b), 3 (c), 4 (d) and 5 (e). Inset: plot of thermal variation of χM−1 for the respective compound. | ||
| Compound | T (K) | C (cm3 K mol−1) | θ (K) |
|---|---|---|---|
| 1 | 2–300 | 2.938 | −0.928 |
| 2 | 2–300 | 1.239 | 0.040 |
| 3 | 20–300 | 0.4312 | −2.507 |
| 4 | 2–300 | 3.095 | −14.407 |
| 5 | 2–300 | 1.094 | 2.653 |
For 1, the χMT value at 300 K is 3.043 cm3 K mol−1 (4.934 μB; Fig. 5a), which is slightly higher than the expected value (2.813 cm3 K mol−1, 4.744 μB) of one and a half isolated spin-only Co(II) ions (S = 3/2, g = 2.0). As T is lowered, χMT decreases continuously to a value of 2.410 cm3 K mol−1 at 2 K, which is attributed to the zero-field splitting (ZFS).
For 2, the χMT value at 300 K is 1.295 cm3 K mol−1 (3.219 μB; Fig. 5b), which is higher than the expected value (1.000 cm3 K mol−1, 2.828 μB) of one isolated spin-only Ni(II) ion (S = 1, g = 2.0). As T is lowered, χMT continuously decreases and reaches a local minimum of 1.246 cm3 K mol−1 at 55.9 K, and then increases to a value 1.273 cm3 K mol−1 at 14.3 K, before dropping quickly to 1.117 cm3 K mol−1 at 2 K. The magnetic behavior of 2 is unusual and interesting, indicative of a strong antiferromagnetic interaction admixture with a very weak ferromagnetic interaction.34
For 3, the χMT value at 300 K is 0.425 cm3 K mol−1 (1.844 μB; Fig. 5c), which is slightly higher than the expected value (0.375 cm3 K mol−1, 1.732 μB) of one isolated spin-only Cu(II) ion (S = 1/2, g = 2.0). Upon cooling, the χMT value decreases monotonically and tends to zero at low temperature (the χMT value of 0.020 cm3 K mol−1 at 2 K), while the χM value increases to a rounded maximum of 0.018 cm3 K mol−1 at 12.4 K and then drops rapidly. These features suggest a dominant antiferromagnetic coupling between the Cu(II) ions. Because the dinuclear copper clusters are interconnected by large Hbtc and Htpim ligands, the overall antiferromagnetic interaction should be attributed to the magnetic exchange coupling within the dinuclear copper cluster.
For 4, the χMT value at 300 K is 2.948 cm3 K mol−1 (4.856 μB; Fig. 5d), which is much higher than the expected value (1.875 cm3 K mol−1, 3.873 μB) of one isolated spin-only Co(II) ion (S = 3/2, g = 2.0), which can be ascribed to the strong orbital contribution to the magnetic moment of Co(II) centers.35 As T is lowered, χMT decreases continuously to a value of 1.801 cm3 K mol−1 at 2 K, which is attributed to the zero-field splitting (ZFS).
For 5, the χMT value at 300 K is 1.148 cm3 K mol−1 (3.031 μB; Fig. 5e), which is slightly higher than the expected value (1.000 cm3 K mol−1, 2.828 μB) of one isolated spin-only Ni(II) ion (S = 1, g = 2.0). The χMT value increases gradually as the temperature is lowed and reaches a maximum value at 9.5 K (1.249 cm3 K mol−1). Below the temperature, the χMT product decreases sharply to 0.816 cm3 K mol−1 at 2 K. The high temperature regime is observed for paramagnetic systems that exhibit dominating ferromagnetic interactions while the χMT decrease at low temperature is usually the signature of weak antiferromagnetic interaction between Ni(II) metal ions and/or zero-field splitting (ZFS) effect.
Conclusions
In summary, five novel coordination polymers based on three large trigonal N-containing ligands (tib, Htpim and pytpy) and polycarboxylate ligands have been successfully synthesized and characterized. These compounds show different fascinating entangled networks. The structural diversities of them indicate that trigonal N-donor ligands play an important role in modulating the entangled modes of coordination polymers. Therefore, the successful isolation of these solid materials not only provides novel examples in the realm of entanglement, but also confirms the introduction of suitable trigonal N-donor ligands may provide a promising approach to constructing new entangled structures. Further studies in this respect are underway in this laboratory.Acknowledgements
This work was financially supported by the NSFC (21571149), the Program for Chongqing Excellent Talents in University, the Fundamental Research Funds for the Central Universities (XDJK2013A027), the Open Fund of Key Laboratory of Synthetic and Natural Functional Molecule Chemistry of the Ministry of Education (338080045), and the Open Fund of Large-Scale Instruments of Southwest University (2012008).Notes and references
-
(a) B. L. Chen, S. Q. Ma, F. Zapata, F. R. Fronczek, E. B. Lobkovsky and H. C. Zhou, Inorg. Chem., 2007, 46, 1233 CrossRef CAS PubMed
; (b) J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC
-
(a) X. Y. Wang, Z. M. Wang and S. Gao, Chem. Commun., 2007, 1127 RSC
-
(a) C. D. Wu, A. Hu, L. Zhang and W. B. Lin, J. Am. Chem. Soc., 2005, 127, 8940 CrossRef CAS PubMed
-
(a) T. Tachikawa, J. R. Choi, M. Fujitsuka and T. Majima, J. Phys. Chem. C, 2008, 112, 14090 CrossRef CAS
-
(a) M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef CAS PubMed
-
(a) S. R. Batten and R. Robson, Angew. Chem., Int. Ed., 1998, 37, 1460 CrossRef
-
(a) S. Hu, A. J. Zhou, Y. H. Zhang, S. Ding and M. L. Tong, Cryst. Growth Des., 2006, 6, 2543 CrossRef CAS
-
(a) E. C. Yang, H. K. Zhao, B. Ding, X. G. Wang and X. J. Zhao, Cryst. Growth Des., 2007, 7, 2009 CrossRef CAS
-
(a) H. Y. Chen, D. R. Xiao, J. Yang, S. W. Yan, J. H. He, X. Wang, Z. L. Ye and E. B. Wang, Inorg. Chem. Commun., 2012, 20, 157 CrossRef CAS
-
(a) R. Saha, B. Joarder, A. S. Roy, S. M. Islam and S. Kumar, Chem.–Eur. J., 2013, 19, 16607 CrossRef CAS PubMed
- X. L. Wang, C. Qin, E. B. Wang, Y. G. Li, Z. M. Su, L. Xu and L. Carlucci, Angew. Chem., Int. Ed., 2005, 44, 5824 CrossRef CAS PubMed
-
(a) T. L. Hennigar, D. C. MacQuarrie, P. Losier, R. D. Rogers and M. J. Zaworotko, Angew. Chem., Int. Ed., 1997, 36, 972 CrossRef CAS
- H. Wu, J. F. Ma, Y. Y. Liu, J. Yang and H. Y. Liu, CrystEngComm, 2011, 13, 7121 RSC
- S. K. Ghosh, J. Ribas and P. K. Bharadwaj, Cryst. Growth Des., 2005, 5, 623 CAS
- I. H. Hwang, J. M. Bae, W. S. Kim, Y. D. Jo, C. Kim, Y. Kim, S. J. Kim and S. Huh, Dalton Trans., 2012, 41, 12759 RSC
- D. R. Xiao, H. Y. Chen, D. Z. Sun, J. H. He, S. W. Yan, J. Yang, X. Wang, R. Yuan and E. B. Wang, CrystEngComm, 2012, 14, 2849 RSC
- W. Q. Kan, J. F. Ma, B. Liu and J. Yang, CrystEngComm, 2012, 14, 286 RSC
-
(a) Y. Rachuri, K. K. Bisht and E. Suresh, Cryst. Growth Des., 2014, 14, 3300 CrossRef CAS
- S. Mukherjee, D. Samanta and P. S. Mukherjee, Cryst. Growth Des., 2013, 13, 5335 CAS
- H. Y. Chen, D. R. Xiao, L. L. Fan, J. H. He, S. W. Yan, G. J. Zhang, D. Z. Sun, Z. L. Ye, R. Yuan and E. B. Wang, CrystEngComm, 2011, 13, 7098 RSC
- H. Wang, J. Xu, D. S. Zhang, Q. Chen, R. M. Wen, Z. Chang and X. H. Bu, Angew. Chem., Int. Ed., 2015, 54, 5966 CrossRef CAS PubMed
- J. Fan, L. Gan, H. Kawaguchi, W. Y. Sun, K. B. Yu and W. X. Tang, Chem.–Eur. J., 2003, 9, 3965 CrossRef CAS PubMed
- M. V. Proskurnina, N. A. Lozinskaya, S. E. Tkachenko and N. S. Zefirov, Russ. J. Org. Chem., 2002, 38, 1149 CrossRef CAS
- C. Liu, Y. B. Ding, X. H. Shi, D. Zhang, M. H. Hu, Y. G. Yin and D. Li, Cryst. Growth Des., 2009, 9, 1275 CAS
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS
-
(a) L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786 CrossRef CAS
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed
- V. A. Blatov, A. P. Shevchenko and V. N. Serezhkin, J. Appl. Crystallogr., 2000, 33, 1193 CrossRef CAS
-
(a) G. H. Wang, Z. G. Li, H. Q. Jia, N. H. Hu and J. W. Xu, Cryst. Growth Des., 2008, 8, 1932 CrossRef CAS
- X. Q. Liang, X. H. Zhou, C. Chen, H. P. Xiao, Y. Z. Li, J. L. Zuo and X. Z. You, Cryst. Growth Des., 2009, 9, 1041 CAS
- C. M. Yuan and X. J. Yang, Inorg. Chem. Commun., 2014, 48, 57 CrossRef CAS
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS
- W. Q. Kan, J. Yang, Y. Y. Liu and J. F. Ma, CrystEngComm, 2012, 14, 6934 RSC
- L. Schlapbach and A. Zuttel, Nature, 2001, 414, 353 CrossRef CAS PubMed
- H. Y. Chen, D. R. Xiao, J. H. He, Z. F. Li, G. J. Zhang, D. Z. Sun, R. Yuan, E. B. Wang and Q. L. Luo, CrystEngComm, 2011, 13, 4988 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Additional structural figures and tables, XRPD patterns, TG-DSC curves, IR spectrum for compounds 1–5. CCDC 1407249–1407251 for 1–3 and 1415509–1415510 for 4–5. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra19086j |
| This journal is © The Royal Society of Chemistry 2016 |