
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A triphenylene-based small molecule compatibiliser using incompatible pendent chains†
Antoine J.
Herbaut
and
Etienne
Baranoff
*
School of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: e.baranoff@bham.ac.uk; Tel: +44 (0)1214142527
First published on 20th January 2016
Abstract
To demonstrate that incompatible pendent chains can be used as a strategy to control the morphology of blends of immiscible materials, we have developed a novel triphenylene-based amphiphile-like mesogen with hydrophobic (alkyl) chains and hydrophilic (2-(2-methoxyethoxy)ethoxy) pendent chains, named TP6Gall hereafter. TP6Gall is a room-temperature liquid crystal presenting a cubic phase with a clearing point of 30 °C. Blends of TP6Gall in various amounts with an equimolar mixture of the hydrophobic 2,3,6,7,10,11-hexahexyloxytriphenylene (TP6) and hydrophilic 2,3,6,7,10,11-hexa(2-(2-methoxyethoxy)ethoxy)triphenylene (TP6EO2M) have been studied by polarised optical microscopy (POM), differential scanning calorimetry (DSC) and small-angle X-ray scattering (SAXS). Without TP6Gall, the TP6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TP6EO2M mixture exhibits large incompatible domains of pure TP6 and pure TP6EO2M. As the quantity of TP6Gall increases in the blend, the size of the domains decreases significantly. Ultimately, when the proportion of TP6Gall reaches 50 mol% of the blend, μm-featured interpenetrated networks of crystalline TP6EO2M and of TP6 mixed with TP6Gall are obtained. Interestingly, a single liquid phase is obtained above the clearing point of the blend. Furthermore, no macrophase separation is observed upon multiple temperature cycles between room-temperature and the temperature above the clearing point of the blend and the interpenetrated network is reliably reformed upon cooling.
:TP6EO2M mixture exhibits large incompatible domains of pure TP6 and pure TP6EO2M. As the quantity of TP6Gall increases in the blend, the size of the domains decreases significantly. Ultimately, when the proportion of TP6Gall reaches 50 mol% of the blend, μm-featured interpenetrated networks of crystalline TP6EO2M and of TP6 mixed with TP6Gall are obtained. Interestingly, a single liquid phase is obtained above the clearing point of the blend. Furthermore, no macrophase separation is observed upon multiple temperature cycles between room-temperature and the temperature above the clearing point of the blend and the interpenetrated network is reliably reformed upon cooling.
1 Introduction
Mixing two compounds is an effective strategy to create new materials combining the properties of the two components and possibly displaying new and enhanced qualities. However when two immiscible substances are mixed together, macrophase separation into two thermodynamically different phases generally occurs and leads to materials with inferior properties. In polymer science, diblock copolymers have been successfully used as compatibilisers to suppress the macrophase separation of polymer blends.1–6 This approach is very important for the whole plastics industry,7 including plastic electronics such as polymer solar cells in which compatibilisers help to control the morphology of donor/acceptor blends and develop more stable devices.4,5Small molecules have well-defined structures and can be sublimed for purification and device preparation, which are advantages over polymers. However, the morphology of blends of functional small molecules is challenging to control; it relies mainly on the use of non-functional additives and/or annealing treatments.8,9 Furthermore, the morphology obtained with such methods is only kinetically trapped and macrophase separation, the thermodynamically stable morphology, will occur over time. In addition, as the role of the additive is purely structural, only a small amount should be used, and as it often remains in the blends, it can impact further the long-term stability of electronic devices.10
To overcome the aforementioned limitations of structural additives, functional small molecule compatibilisers are expected to be a viable strategy to control the morphology of blends of small molecules in a simple manner, yet examples in the literature are scarce. Kim et al. have developed thiophene–C60 dyads as interfacial agents for compatibilising blends of P3HT and PCBM for organic solar cell.11 They have shown that macrophase separation and domain coarsening can be suppressed, which result in increased efficiency and stability. A similar approach was recently reported by Raja et al.12
In another domain of application, Date and Bruce have prepared a rod-disk shape amphiphile to suppress the macrophase separation in a blend of rod and disklike mesogens.13 A homogenous nematic phase was obtained over a large range of amphiphile concentration demonstrating the compatibilisation of the rod and disk-like mesogens. Kouwer and Mehl also developed a series of mesogenic rod-disk shape amphiphiles.14 Mixing the amphiphile with the rod-like mesogen shows a homogeneous isotropic phase over a wide range of temperature while mixing with the disklike mesogen shows one monotropic nematic phase. No mixing studies were carried out on the mixture of rods, disks, and amphiphiles. Other amphiphile-like mesogens were described in the literature but no studies of blends were carried out.15–19
In these examples, the cores of the molecules are acting as the incompatible parts mainly because of their shape (rod/sphere and rod/disk), which can greatly limit the available molecular designs for the concomitant optimisation of the morphology of the blend and of the sought after materials properties.
In order to demonstrate a more general approach, we have explored a strategy based on incompatible peripheral chains: the pendent chains would act as the immiscible elements while the core of the molecule would provide the function of interest. Herein, as a model study, we report a novel amphiphile-like mesogen, TP6Gall, and its use as a compatibilising agent to stabilise blends of hydrophobic (TP6, 2,3,6,7,10,11-hexahexyloxytriphenylene) and hydrophilic (TP6EO2M, 2,3,6,7,10,11-hexa(1,4,7-trioxaoctyl)triphenylene) triphenylenes, Scheme 1. We have chosen triphenylenes as the model aromatic cores because they are relatively simple to synthesise and are a well-known class of discotic liquid crystals.20 In this model system, the opportunity for mesophases (ordered molecules in the bulk) enables easier characterisation of bulk morphology compared to amorphous materials.
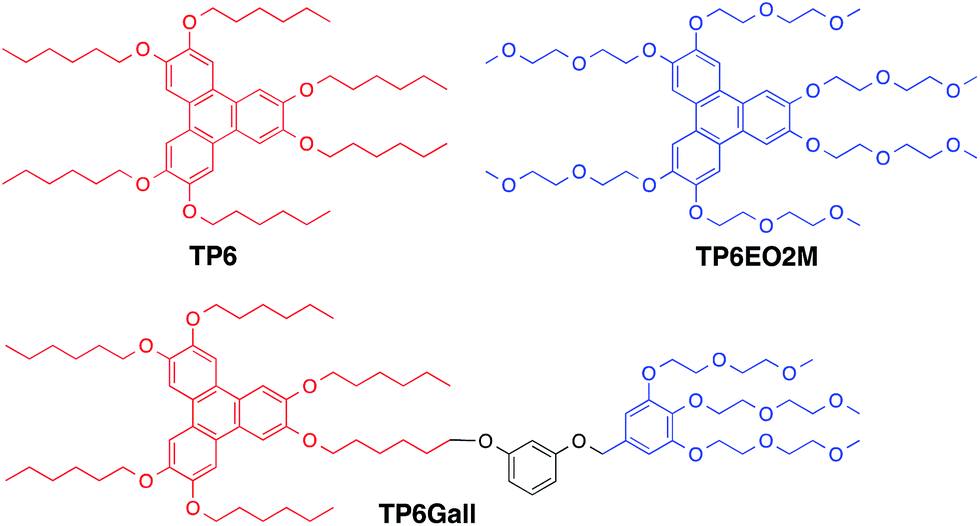 | ||
| Scheme 1 Chemical structure of the investigated compounds. | ||
Blends of TP6Gall (0–50 mol%) with an equimolar TP6:TP6EO2M mixture have been studied by polarised optical microscopy (POM), differential scanning calorimetry (DSC) and small-angle X-ray scattering (SAXS). While the TP6:TP6EO2M mixture results in large incompatible domains of pure TP6 and pure TP6EO2M, addition of TP6Gall leads to smaller domains as the quantity of TP6Gall increases. With 50 mol% TP6Gall, μm-featured interpenetrated networks of crystalline TP6EO2M and of TP6/TP6Gall mixture are obtained. Interestingly, a single liquid phase is obtained above the clearing point of the blend and no macrophase separation is observed upon multiple temperature cycles between RT and temperature above the clearing point of the blend: the interpenetrated network is reliably reformed upon cooling.
2 Results and discussion
2.1 Synthesis
TP6 and TP6EO2M were obtained accordingly to literature procedures21 with minor modifications.22 In particular TP6EO2M necessitated purification by HPLC to be obtained pure as minor triphenylene-based impurities could not be fully removed by chromatography columns.22TP6Gall was obtained in 12 steps with 5% overall yield using a convergent synthesis to couple the hydrophilic and the hydrophobic fragments, Scheme 2. The hydrophobic triphenylene 16 was synthesized by the biphenyl–phenyl coupling method.23 Briefly, a FeCl3-based Scholl type oxidative coupling between 3,3′,4,4′-tetrakis(hexyloxy)-1,1′-biphenyl and 1-(hexyloxy)-2-isopropoxybenzene afforded the monohydroxy-pentahexyloxytriphenylene 11,24 which was reacted with 1,6-dibromohexane. The hydrophilic moiety 14 was derived from ethyl gallate substituted with 2-(2-methoxyethoxy)ethanol chains, which ester function was reduced to alcohol and then chlorinated. 1,3-Dihydroxybenzene was used as a linker between the hydrophilic and the hydrophobic parts (synthetic details in ESI†).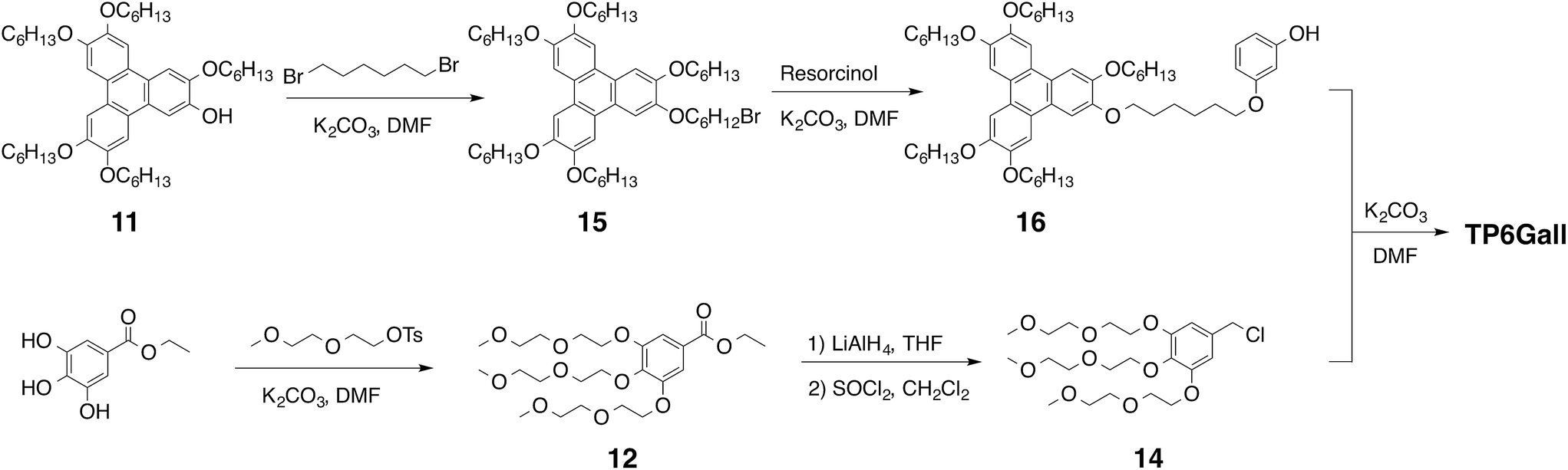 | ||
| Scheme 2 Synthetic strategy for TP6Gall. | ||
2.2 Thermal properties of pure compounds
As reported in the literature, TP6 exhibits a columnar hexagonal liquid crystalline phase from 69 °C to 99 °C and TP6EO2M shows only a Cr–Iso transition at 52 °C.21,25TP6Gall is liquid crystalline at room temperature with a clearing point at 30 °C. X-ray diffraction profile of TP6Gall shows three peaks (Fig. 1), which follow a Q-ratio of 2:√5:√6 (indexed as (200), (210), and (211)), indicative of a cubic lattice with a lattice parameter a = 33.7 Å, which can be ascribed to an extended form of TP6Gall (∼35 Å, inset Fig. 1) with some interpenetration of the pendent chains.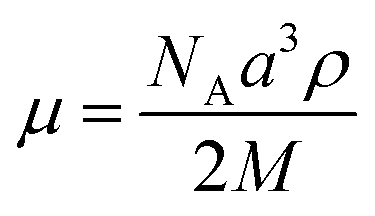 | (1) |
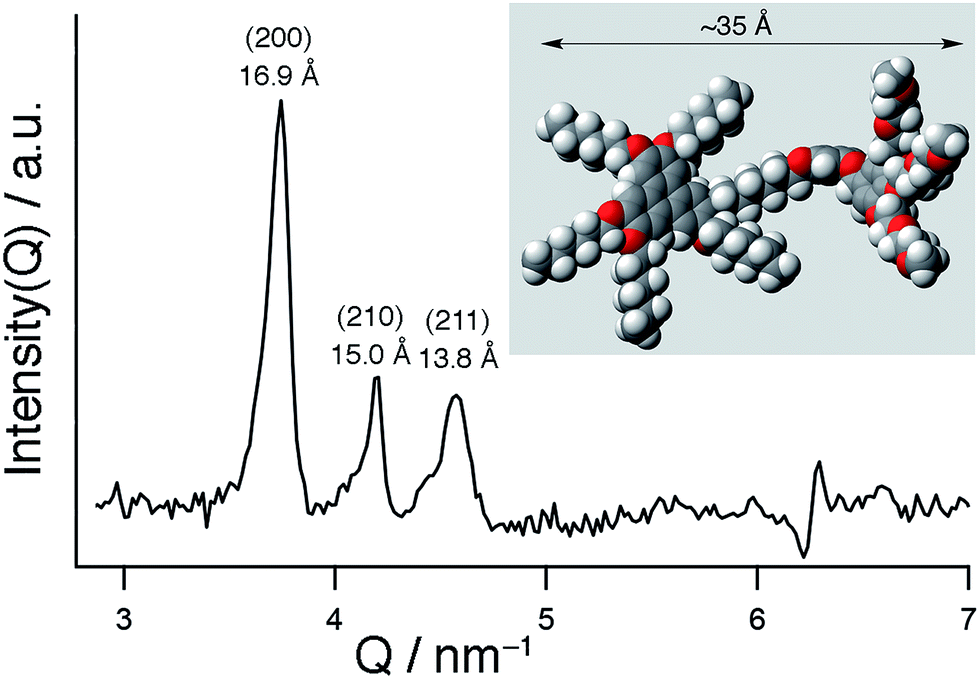 | ||
| Fig. 1 SAXS profile of TP6Gall at 25 °C. Inset: MM2 model of TP6Gall. | ||
Using eqn (1) to calculate μ, the number of molecules per micelle for a cubic centred lattice, a value of about 10 molecules per micelle is obtained. With an interdisc distance of ∼3.6 Å,26 the length of the micelle would be 32.4 Å, in line with the lattice parameter. Therefore we attribute the mesophase of TP6Gall to a centred cubic phase.
When TP6 and TP6EO2M are mixed in an equimolar ratio,‡ macrophase separation is observed and the thermal behaviour of each phase is similar to the individual components (Table 1). The POM clearly demonstrates the immiscibility of the two triphenylenes in the isotropic phase (Fig. 2a). Indeed, two immiscible liquid phases are observed. Upon cooling this blend, the isotropic TP6 domain becomes liquid crystalline around 70 °C (Fig. 2b) then crystalline at ∼50 °C. At ∼40 °C the isotropic TP6EO2M domain also becomes crystalline (Fig. 2c). This behaviour is further supported by XRD analysis (Fig. 3). The SAXS profile of the blend at 80 °C (Fig. 3d) shows the main peak corresponding to TP6 in the hexagonal columnar phase (Fig. 3a). At 60 °C (Fig. 3e) the SAXS profile shows the pattern for TP6 in the crystalline phase alone (Fig. 3b), while at 25 °C (Fig. 2f) the SAXS pattern is the combination of the patterns for crystalline TP6 (Fig. 3b) and crystalline TP6EO2M (Fig. 3c).
| Sample | 2nd heating | Transition |
|---|---|---|
|
a From POM.
b 50:50 mol%.
c In mol%. Cr: crystal, Iso: isotropic, Cub: cubic, Colh: columnar hexagonal, M: unknown mesophase, X: unknown phase.
|
||
| TP6 | 69 (46) | Cr–Colh |
| 99 (6) | Colh–Iso | |
| TP6EO2M | 52a | Cr–Iso |
| TP6Gall | 30 (19) | Cub–Iso |
|
TP6:TP6Gallb |
39 (8) | M–M |
| 49 (9) | M–Iso | |
|
TP6:TP6EO2Mb |
51 (21) | Cr–Iso (TP6EO2M) |
| 66 (17) | Cr–Colh (TP6) | |
| 88a | Colh–Iso (TP6) | |
|
||
(TP6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :TP6EO2M):
TP6Gall
c :TP6EO2M):
TP6Gall
c
|
||
| 95:5 |
52 (19) | Cr–Iso (TP6EO2M) |
| 66 (16) | Cr–Colh (TP6) | |
| 85 a | Colh–Iso (TP6) | |
| 80:20 |
40 | X–X |
| 50 | X–X | |
| 53 | X–X | |
| 63 | X–Iso | |
| 50:50 |
41 | M + Cr–X + Cr |
| 48 | X + Cr–Iso | |
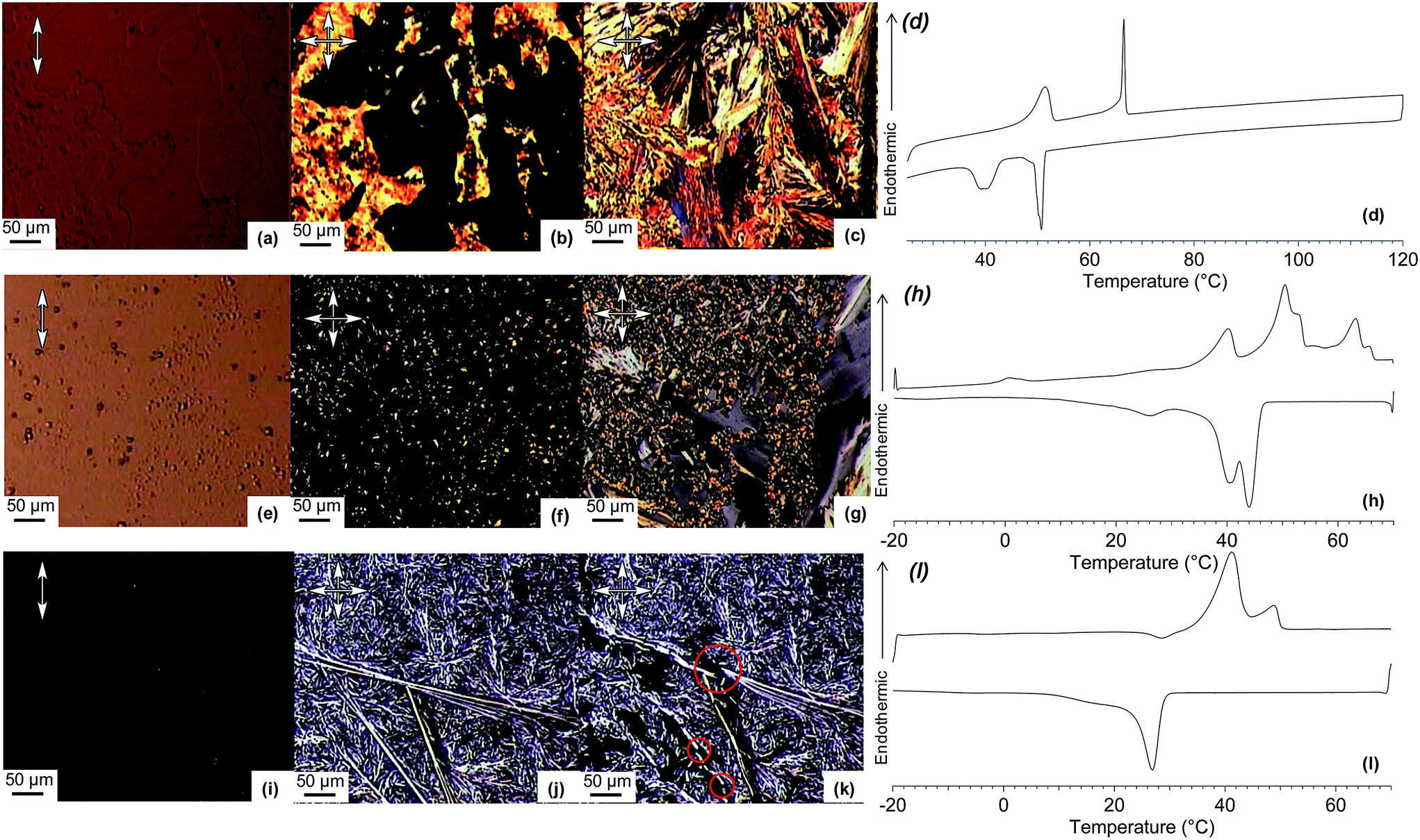 | ||
| Fig. 2
TP6:TP6EO2M equimolar mixtures. Top: with 0 mol% TP6Gall (a) POM image at 90 °C; (b) at 50 °C; (c) at 25 °C; (d) DSC curves of second heating and second cooling. Middle: with 20 mol% TP6Gall (e) POM image at 70 °C; (f) at 50 °C; (g) at 25 °C; (h) DSC curves of second heating and second cooling. Bottom: with 50 mol% TP6Gall (i) POM image at 70 °C; (j) at 50 °C; (k) at 25 °C; (l) DSC curves of second heating and second cooling (10 °C min−1). Single arrow: parallel polarisers, crossed arrows: crossed polarisers. | ||
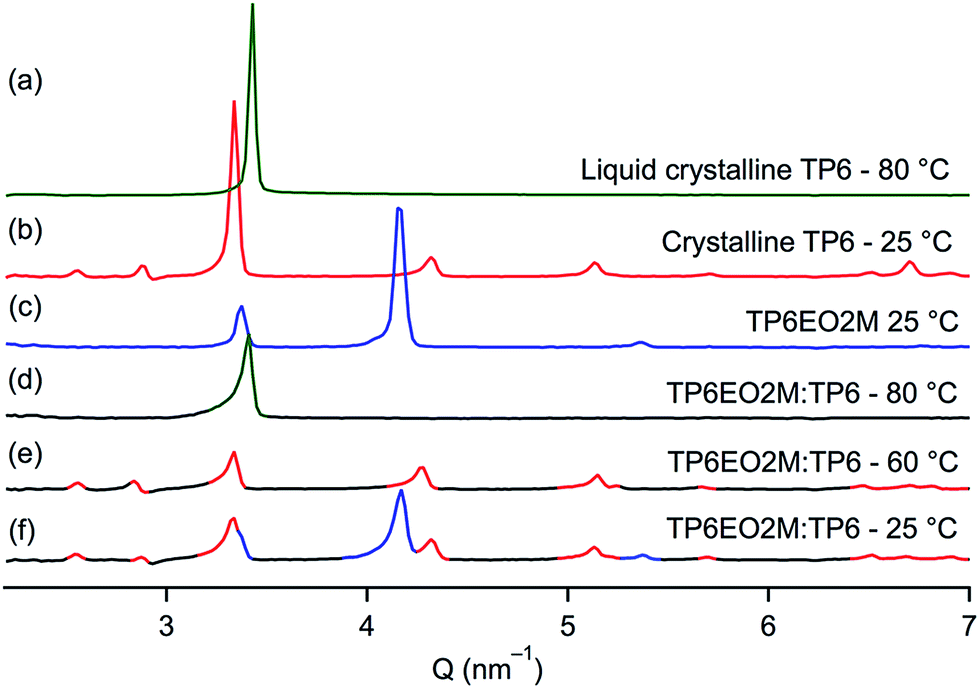 | ||
| Fig. 3 SAXS patterns of (a) TP6 at 80 °C (LC phase), (b) TP6 at 25 °C (crystal phase), (c) TP6EO2M at 25 °C (crystal phase) and a TP6:TP6EO2M 50 mol%:50 mol% mixture at (d) 80 °C, (e) 60 °C, (f) 25 °C. | ||
Upon addition of TP6Gall to the TP6:TP6EO2M mix, the behaviour and morphology of the blend is altered. With only a small amount (5 mol%) of TP6Gall, the blend behaves similarly to the non-doped mixture, yet smaller domains of TP6 and TP6EO2M can be observed on POM (Fig. S12†) pointing to an impact of the compatibiliser on the morphology of the blend already at low concentration. DSC curves (Fig. S13†) and SAXS patterns (Fig. S14†) do not differ much from the TP6:TP6EO2M equimolar blend. No diffraction pattern or transition temperature corresponding to pure TP6Gall was observed. While it would be convenient to assume that TP6Gall is well dispersed at the TP6/TP6EO2M interface, TP6Gall is more likely well mixed within a TP6-rich domain. Indeed when TP6 and TP6Gall are mixed in an equimolar ratio, homogeneous mesophases and isotropic phase are observed (Fig. S21† and SAXS pattern Fig. 4a), demonstrating that TP6 and TP6Gall are miscible.
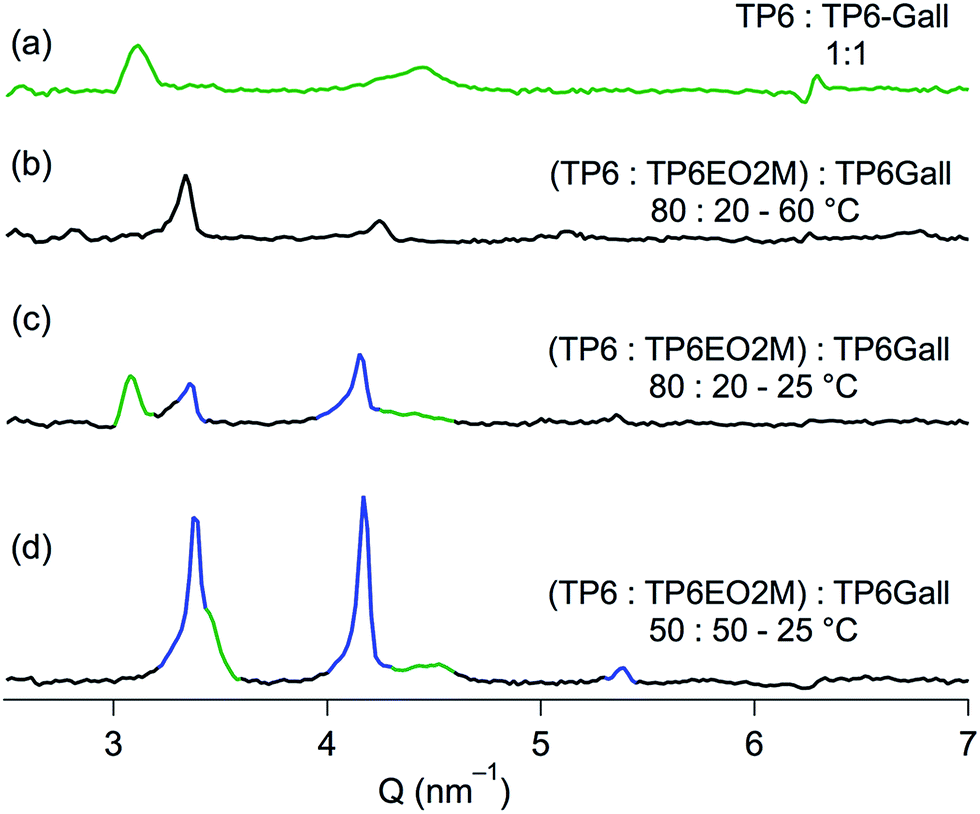 | ||
| Fig. 4 SAXS pattern of (a) TP6:TP6Gall equimolar mixture at 25 °C and (TP6:TP6EO2M):TP6Gall mixture (b) with 20 mol% TP6Gall at 60 °C, (c) with 20 mol% TP6Gall at 25 °C, (d) with 50 mol% TP6Gall at 25 °C. | ||
Significant changes were observed with 20 mol% TP6Gall. Importantly, the size of the domains appears again smaller than in the TP6:TP6EO2M mix (Fig. 2a–c) and blends containing 5 mol% TP6Gall (Fig. S12†). Complex textures observed in POM (Fig. 2e–g) and overlapping phase transitions in DSC (Fig. 2h) preclude detailed analysis of this blend. Nevertheless, mainly two domains can be observed by POM at all temperature, including after the clearing point (63 °C) with two immiscible liquid phases. Upon cooling, one of the isotropic domains forms an unknown LC phase (SAXS pattern in Fig. 4b). No definite conclusion can be drawn as the SAXS profile resembles both the profile of crystalline TP6EO2M and the profile of the TP6:TP6Gall mix. Upon further cooling to 25 °C, two domains can be attributed by analysing the SAXS profile (Fig. 4c). First, a crystalline domain is composed of TP6EO2M. Indeed, the peaks at 3.33, 4.15, and 5.39 nm−1 (highlighted in blue in Fig. 4c) are similar to the three peaks observed at 3.37, 4.16, and 5.36 nm−1 for the pure TP6EO2M (Fig. 3c). The composition of the unknown mesophase is ascribed to a TP6:TP6Gall mixture based on the similarity of the peaks at 3.08 and 4.35 (broad) nm−1 (highlighted in green in Fig. 4c), and the pattern observed for the TP6:TP6Gall mixture with peaks at 3.12 and 4.40 (broad) nm−1 (Fig. 4a).
Most interesting is the mixture containing 50 mol% TP6Gall that is with a TP6 25 mol%:TP6Gall 50 mol%:TP6EO2M 25 mol% final composition. The thermal behaviour of this blend looks much simpler than the 20 mol% case. For the first time in this system a single isotropic phase was observed under POM above the clearing point at 48 °C (Fig. 2i). Upon cooling, micro-segregation of a network of crystalline domains in a mesophase was observed (Fig. 1j). The crystalline nature of the network is confirmed as it is readily broken into smaller pieces when applying pressure on the sample (Fig. 2k).§ As for the other mixtures, the crystalline phase is composed of TP6EO2M as seen in the SAXS pattern (Fig. 4d). The unknown mesophase is again attributed to a TP6:TP6Gall mixture based on the SAXS patterns, a relatively narrow peak as 3.49 nm−1 (overlapped with the signal of TP6EO2M) and a broad peak at 4.50 nm−1 (Fig. 3d). Compared to the pure TP6:TP6Gall mixture both peaks are shifted to higher Q values, which is attributed to different TP6/TP6Gall ratio. The DSC traces (Fig. 2l) are similar to the traces observed for the TP6:TP6Gall mixture (Fig. S22†) with two endothermic transitions on heating and two exothermic transitions on cooling. The first endothermic transition at 40 °C has much higher enthalpy than in the TP6:TP6Gall mixture. It is attributed to an overlapping transition involving TP6EO2M.
An important difference with previous blends is the morphology of the present blend, as now TP6EO2M and TP6/TP6Gall form densely interpenetrating networks at the micrometer scale. Furthermore this particular morphology is well reproducible upon heating–cooling cycles, most likely because the liquid phase is a single isotropic phase, even if mechanically challenged during the process.§ On heating, the crystalline and LC domains melt and a homogenous isotropic phase is obtained. On cooling from the isotropic phase, the growing crystals do not aggregate into large domain and stays well dispersed into the liquid crystalline matrix.
3 Conclusions
In conclusion, a novel mesogenic amphiphile, TP6Gall, was synthesized and its compatibilising behaviour was studied. TP6Gall shows a cubic liquid crystalline lattice at room temperature. The blend of two incompatible triphenylenes, the hydrophobic TP6 and the hydrophilic TP6EO2M, shows macrophase separation and a biphasic liquid phase is obtained upon heating. On addition of small amount of TP6Gall smaller domains are observed but macrophase separation still occurs. At higher concentration (50 mol%), TP6 and TP6EO2M micro segregate into a network of crystalline TP6EO2M in a TP6:TP6Gall liquid crystalline matrix. Notably, the three-component melt is a single isotropic phase and the micro-segregated network is reliably formed during cooling. We have therefore demonstrated compatibilisation of two immiscible triphenylenes using TP6Gall. Uniquely we have developed a system based on incompatibility of peripheral chains instead of molecular shapes as used in the few published examples of small molecules compatibilisation. This is expected to be advantageous for the development of functional materials blends, as it allows for greater flexibility in the design of the core molecules hence enables simple modification of the properties of the final materials. Importantly, the small molecule compatibiliser acts as a functional structural agent and as such the amount that can be used in the blend does not necessarily need to be small.
4 Experimental
4.1 Materials
Dichloromethane, hexane, ethanol, methanol, N,N′-dimethylformamide (DMF), tetrahydrofurane (THF), acetic acid, and sulfuric acid were purchased from Fisher. Acetone and ethyl acetate were purchased from VWR. Diethyl ether was purchased from Sigma Aldrich. Hydrobromic acid was purchased from Alfa Aesar. All solvents were used without further purification except for ethyl acetate, which was distilled in vacuo. Column chromatography was carried out on silica gel 60 [Alfa Aesar, 0.040–0.063 mm]. Reaction progress was followed with TLC plate on aluminium support (silica: Alugram® SIL G/UV254).4.2 Equipment
NMR spectra were recorded on Brucker Avance III 400 (1H 400 MHz, 13C 101 MHz) instrument and are reported relative to the residual solvent as the internal standard (CDCl3: δ [1H] = 7.26 ppm; δ [13C] = 77.16 ppm; DMSO-d6: δ [1H] = 2.50 ppm; δ [13C] = 39.52 ppm). Coupling constants are expressed in Hz. High resolution mass spectra (HRMS) were determined from Micromass ZABspec mass spectrometer in TOP MS ES+ mode and are reported as (m/z (%)). IR spectra were recorded on Perkin Elmer Spectrum 100 FT-IR spectrometer. Polarized optical microscopy images were recorded on an Olympus C-CMAD-2 device using a JVC TK-C1380 camera. Heating was controlled using a Linkam Scientific LTS 350 apparatus. X-ray diffraction measurements were recorded using a Macor cup on an Anton Paar PANalytical empyrean X-ray diffractometer equipped with a CuKα X-ray source (λ = 1.540598 Å). DSC curves were recorded on a Perkin Elmer DSC 7 device (for ambient to high temperature) or on a Mettler Toledo DSC 1 STARe System (for sub-ambient to high temperature). All DSC results shown are taken during a second heating–cooling cycle. Transition temperatures are taken at the maximum transition peak.4.3 Material synthesis
:3) to afford 12 as a colourless oil (486 mg, 49%). 1H NMR (400 MHz, CDCl3) δ 7.28 (s, 2H, ArCH), 4.33 (q, J = 7.1 Hz, 2H, –CH2OCOR), 4.26–4.17 (m, 6H, –CH2–), 3.89–3.84 (m, 4H, –CH2–), 3.82–3.78 (m, 2H, –CH2–), 3.74–3.68 (m, 6H, –CH2–), 3.58–3.50 (m, 6H, –CH2–), 3.37 (s, 6H, –CH2–), 3.36 (s, 3H, –CH3), 1.36 (t, J = 7.1 Hz, 3H, CH3CH2OCOR). 13C NMR (101 MHz, CDCl3) δ 166.2 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 152.4 (2 ArC), 142.6 (ArC), 125.5 (ArC) 109.1 (2 ArCH), 72.6 (–CH2–), 72.2 (–CH2–), 72.1 (2 –CH2–), 70.9 (2 –CH2–), 70.7 (–CH2–), 70.6 (–CH2–), 69.8 (2 –CH2–), 69.0 (2 –CH2–), 61.1 (–CH2OCOR), 59.2 (2 –CH3), 59.1 (–CH3), 14.5 (CH3CH2OCOR). HRMS (TOF ES+): m/z [M + Na]+ = 527.2479 (calculated: 527.2468), [M + K]+ = 543.5.
O), 152.4 (2 ArC), 142.6 (ArC), 125.5 (ArC) 109.1 (2 ArCH), 72.6 (–CH2–), 72.2 (–CH2–), 72.1 (2 –CH2–), 70.9 (2 –CH2–), 70.7 (–CH2–), 70.6 (–CH2–), 69.8 (2 –CH2–), 69.0 (2 –CH2–), 61.1 (–CH2OCOR), 59.2 (2 –CH3), 59.1 (–CH3), 14.5 (CH3CH2OCOR). HRMS (TOF ES+): m/z [M + Na]+ = 527.2479 (calculated: 527.2468), [M + K]+ = 543.5.
To a solution of 3,4,5-tris(1,4,7-trioxaoctyl)benzyl alcohol (200 mg, 0.43 mmol, 1 equiv.) in dry DCM (25 mL) was added dropwise a solution of SOCl2 (320 μL, 4.3 mmol, 10 equiv.) in dry DCM (10 mL) at room temperature. The solution was stirred at room temperature for 3 h. The solvent was evaporated in vacuo, affording 14 in a quantitative yield. 1H NMR (400 MHz, CDCl3) δ 6.62 (s, 2H, ArCH), 4.48 (s, 2H, Ar–CH2–Cl), 4.21–4.12 (m, 6H, Ar–O–CH2–), 3.89–3.83 (m, 4H, –CH2–), 3.82–3.77 (m, 2H, –CH2–), 3.74–3.69 (m, 6H, –CH2–), 3.60–3.52 (m, 6H, –CH2–), 3.38 (s, 6H, –CH3), 3.38 (s, 3H, –CH3). 13C NMR (101 MHz, CDCl3) δ 152.8 (3 ArC), 132.9 (ArC), 108.4 (2 ArCH), 72.5 (–CH2–), 72.2 (–CH2–), 72.2 (2 –CH2–), 70.9 (2 –CH2–), 70.7 (–CH2–), 70.6 (–CH2–), 69.9 (2 –CH2–), 69.1 (2 –CH2–), 59.2 (3 –CH3), 46.7 (Ar–CH2–Cl). HRMS (TOF ES+): m/z [M + Na]+ = 503.2005 (calculated: 503.2024). IR: (neat)/cm−1 2925, 2876, 2823; 1591, 1504 (aromatic ring stretch); 1333 (aromatic ether φ–O–C); 1101 (aliphatic ether).
:6). After 20 h stirring, the reaction mixture was cooled down to room temperature and acidified with a 1 M aq. solution of HCl. Extraction with ethyl acetate followed by evaporation in vacuo of the solvent afforded a orange solid which was purified by column chromatography (SiO2, DCM). 016 was obtained as a white solid (105.9 mg, 42%). 1H NMR (400 MHz, CDCl3) δ 7.84 (s, 6H, ArCH), 7.10 (t, J = 8.4 Hz, 1H, ArCH), 6.50–6.44 (m, 1H, ArCH), 6.43–6.37 (m, 2H, ArCH), 4.99 (s, 1H, –OH), 4.23 (t, J = 6.6 Hz, 12H, –OCH2), 3.95 (t, J = 6.4 Hz, 2H, –OCH2), 2.02–1.88 (m, 12H, –CH2–), 1.88–1.77 (m, 2H, –CH2–), 1.67–1.51 (m, 14H, –CH2–), 1.47–1.32 (m, 20H, –CH2–), 1.01–0.86 (m, 15H, –CH3). 13C NMR (101 MHz, CDCl3) δ 160.6, 156.9, 149.2, 149.1, 149.1, 149.1, 130.2, 123.9, 123.9, 123.8, 107.8, 107.6, 107.1, 102.2, 69.9, 68.0, 31.8, 29.6, 29.3, 26.1, 26.0, 14.2. HRMS (TOF ES+): m/z [M] = 936.6451 (calculated: 936.6479).
Acknowledgements
We thank the School of Metallurgy and Materials (University of Birmingham) for access to its analytical facility for DSC studies and Dr Martin Hollamby for helpful discussions. AJH thanks the School of Chemistry, University of Birmingham, for a studentship, and the EPSRC for underpinning support.Notes and references
- R. M. Ho, Y. W. Chiang, C. C. Lin and S. J. Bai, Macromolecules, 2002, 35, 1299 CrossRef CAS.
- P. Jannasch, H. Hassander and B. Wesslen, J. Polym. Sci., Part B: Polym. Phys., 1996, 34, 1289 CrossRef CAS.
- H. J. Kim, J. H. Kim, J. H. Ryu, Y. Kim, H. Kang, W. B. Lee, T. S. Kim and B. J. Kim, ACS Nano, 2014, 8, 10461 CrossRef CAS PubMed.
- S. Rajaram, P. B. Armstrong, B. J. Kim and J. M. J. Frechet, Chem. Mater., 2009, 21, 1775 CrossRef CAS.
- K. Sivula, Z. T. Ball, N. Watanabe and J. M. J. Fréchet, Adv. Mater., 2006, 18, 206 CrossRef CAS.
- L. A. Utracki, Can. J. Chem. Eng., 2002, 80, 1008 CrossRef CAS.
- J. Markarian, Plast. Addit. Compd., 2004, 6, 22 Search PubMed.
- G. De Luca, W. Pisula, D. Credgington, E. Treossi, O. Fenwick, G. M. Lazzerini, R. Dabirian, E. Orgiu, A. Liscio, V. Palermo, K. Mullen, F. Cacialli and P. Samori, Adv. Funct. Mater., 2011, 21, 1279 CrossRef CAS.
- Y. Diao, L. Shaw, Z. A. Bao and S. C. B. Mannsfeld, Energy Environ. Sci., 2014, 7, 2145 CAS.
- W. Kim, J. K. Kim, E. Kim, T. K. Ahn, D. H. Wang and J. H. Park, J. Phys. Chem. C, 2015, 119, 5954 CAS.
- J. B. Kim, K. Allen, S. J. Oh, S. Lee, M. F. Toney, Y. S. Kim, C. R. Kagan, C. Nuckolls and Y.-L. Loo, Chem. Mater., 2010, 22, 5762 CrossRef CAS.
- R. Raja, W. S. Liu, C. Y. Hsiow, Y. J. Hsieh, S. P. Rwei, W. Y. Chiu and L. Wang, Org. Electron., 2014, 15, 2223 CrossRef CAS.
- R. W. Date and D. W. Bruce, J. Am. Chem. Soc., 2003, 125, 9012 CrossRef CAS PubMed.
- P. H. J. Kouwer and G. H. Mehl, J. Mater. Chem., 2009, 19, 1564 RSC.
- J. J. Hunt, R. W. Date, B. A. Timimi, G. R. Luckhurst and D. W. Bruce, J. Am. Chem. Soc., 2001, 123, 10115 CrossRef CAS PubMed.
- C. T. Imrie, Z. Lu, S. J. Picken and Z. Yildirim, Chem. Commun., 2007, 1245 RSC.
- K. U. Jeong, A. J. Jing, B. Mansdorf, M. J. Graham, D. K. Yang, F. W. Harris and S. Z. D. Cheng, Chem. Mater., 2007, 19, 2921 CrossRef CAS.
- J. H. Jung, S.-E. Kim, E. K. Song, K. S. Ha, N. Kim, Y. Cao, C.-C. Tsai, S. Z. D. Cheng, S. H. Lee and K.-U. Jeong, Chem. Mater., 2010, 22, 4798 CrossRef CAS.
- S. Kumar, J. Naidu and S. K. Varshney, Mol. Cryst. Liq. Cryst., 2004, 411, 355 CrossRef.
- S. Kumar, Liq. Cryst., 2004, 31, 1037 CrossRef CAS.
- R. C. Borner, R. J. Bushby and A. N. Cammidge, Liq. Cryst., 2006, 33, 1439 CrossRef CAS.
- A. Herbaut and E. Baranoff, Chimia, 2015, 69, 520 CrossRef CAS PubMed.
- N. Boden, R. J. Bushby and A. N. Cammidge, J. Am. Chem. Soc., 1995, 117, 924 CrossRef CAS.
- R. J. Bushby and Z. B. Lu, Synthesis, 2001, 763 CrossRef CAS.
- N. Terasawa, H. Monobe and K. Kiyohara, J. Fluorine Chem., 2006, 127, 954 CrossRef CAS.
- E. O. Arikainen, N. Boden, R. J. Bushby, J. Clements, B. Movaghar and A. Wood, J. Mater. Chem., 1995, 5, 2161 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Full synthetic details, 1H and 13C NMR spectra, additional POM images, DSC curves, SAXS patterns. See DOI: 10.1039/c5ra23904d |
| ‡ All blends have been prepared by dissolving the components in dichloromethane at the required ratio followed by solvent evaporation and overnight drying under vacuum. Data discussed are obtained after heating at least once the mixtures above the clearing point. |
| § Mechanical pressure applied with a spatula on the glass covering the sample. |
| This journal is © The Royal Society of Chemistry 2016 |