
In situ polymerization of liquid-crystalline thin films of electron-transporting perylene tetracarboxylic bisimide bearing cyclotetrasiloxane rings†
Abstract
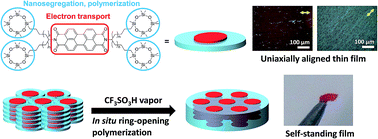
A perylene tetracarboxylic bisimide (PTCBI) derivative bearing four cyclotetrasiloxane rings that forms a columnar phase at room temperature despite the presence of the bulky cyclotetrasiloxane rings was prepared. The electron mobility in the columnar phase of this PTCBI derivative at room temperature is 0.12 cm2 V−1 s−1, which is comparable to those of molecular crystals. The PTCBI derivative is soluble in various organic solvents and liquid crystalline thin films were successfully produced by a spin-coating method. The spin-coated films were insolubilized upon exposure to trifluoromethanesulfonic acid vapor. The columnar structure in the LC phase was retained during polymerization. Using a friction transfer method, macroscopically aligned LC thin films were produced and the optical anisotropy was retained after polymerization.
 Please wait while we load your content...
Please wait while we load your content...

