
Novel benzimidazole–oxadiazole hybrid molecules as promising antimicrobial agents†
N. Shruthia,
Boja Poojary*a,
Vasantha Kumara,
Mumtaz Mohammed Hussainb,
Vaishali M. Raic,
Vinitha R. Paic,
Mahima Bhata and
B. C. Revannasiddappad
aDepartment of Chemistry, Mangalore University, Mangalagangothri-574199, Karnataka, India. E-mail: bojapoojary@gmail.com; Fax: +91-824-2287367; Tel: +91-824-2287262
bDepartment of Pharmaceutical Chemistry, S.J.M. College of Pharmacy, Chitradurga-575501, Karnataka, India
cDepartment of Biochemistry, Yenepoya University, Deralakatta, 575 018, Mangalore, Karnataka, India
dDepartment of Pharmacology, N.G.S.M. Institute of Pharmaceutical Sciences, Paneer, Deralakatta, Mangalore-575 018, Karnataka, India
First published on 6th January 2016
Abstract
In the present study, we describe the design and expeditious synthesis of novel 2-aryl-5-(3-aryl-[1,2,4]-oxadiazol-5-yl)-1-methyl-1H-benzo[d]imidazole hybrid molecules as promising antimicrobial agents. The core moiety 2-aryl-ethyl-1H-benzo[d]imidazole-5-carboxylate was efficiently synthesized by a rapid ‘one pot’ nitro reductive cyclization reaction using sodium dithionite as reagent. All the compounds were screened for their antimicrobial activities and the active compounds were screened for their anti-tubercular activity against Mycobacterium tuberculosis H37Rv strain by the Microplate Alamar Blue Assay method. Compounds 8k, 8n, 8p and 8r exhibited potent anti-tubercular activity with MIC of 1.6 μg mL−1, which is two times more potent than the standard drugs pyrazinamide and ciprofloxacin and fourfold superior to streptomycin and isoniazid. Further, potent compounds were tested for their preliminary toxicity by hemolytic assay, where the compounds remain nontoxic even at higher concentration and showed good selectivity index. The investigation of cytotoxicity against normal embryonic kidney cell line HEK 293 by MTT assay showed IC50 value of more than 355 μg mL−1 for all the tested potent compounds. From the screening study, 8k, 8n, 8p and 8r emerged as strong candidates with potent antimicrobial activities and good ADME parameters. Attempts were also made to establish the structural activity relationships among the tested compounds.
1. Introduction
Antibiotics are the most important weapons in the fight against microbial infections and have enormously benefited the health-related quality of human life since their introduction. However, over the past few decades, these health benefits are under threat as many commonly used antibiotics are becoming less sensitive and less effective against certain illnesses, not only because of toxic reactions, but also due to the emergence of drug-resistant bacteria. After AIDS, tuberculosis (TB) caused by Mycobacterium tuberculosis is the world's second most serious and fatal chronic infectious disease.1 People with weak immune systems, for example, HIV positive, diabetes,2 cancer3 and transplant4 patients, are more susceptible to TB infection. The unique structure and chemical composition of the mycobacterial cell wall prevent successful entry of drugs which renders most antibiotics ineffective making TB treatment very difficult. Furthermore, this problem has become more critical with the emergence of multi-drug-resistant and extensively drug-resistant strains of M. tuberculosis against both the first line as well as second line drugs.5–7 But no such new effective TB drugs have been launched in the market for the past few decades. It is also necessary that effective therapeutic agents should possess good safety profiles, i.e. good selectivity index and less toxicity. But there are many reports found in the literature where the most potent compounds are not effective because of their toxicity. Also, potency and selectivity are extremely sensitive to minor changes in the chemical structure of the drug.8–12 Hence, there is an urgent requirement of developing new antimycobacterial drugs with a unique structure, effective mode of action, with reduced toxicity and good selectivity compared to the existing drugs.The benzimidazole nucleus is considered as ‘Master Key Core’ in many compounds aimed at different targets to bring out varied pharmacological properties. Benzimidazole chemistry has gained prominent importance with the discovery of N-ribosyldimethylbenzimidazole which serves as an axial ligand for cobalt in vitamin B12.13 In the synthesis of benzimidazoles reduction of nitro group followed by cyclization is the main process which conventionally involves two steps. One pot reactions are gaining significant importance to conduct syntheses in a more sustainable fashion with an aim of minimizing reagent and solvent use, as well as to reduce the isolation steps.14 Benzimidazole and its derivatives represent one of the highly important biologically active classes of compounds possessing various biological activities including antiviral,15 anthelmintic,16 anticancer,17,18 antihistamine,19 antihypertensive,20 antineoplastic,21 antiprotozoal,22 and anti-hepatitis B virus activities.23 Only few reports are available of antitubercular activity of benzimidazoles.24–27 These developments sparked active research to explore the nucleus with varied activities. Optimization of substituents around the benzimidazole nucleus has resulted in many drugs like carbendazim (antifungal agent), albendazole, mebendazole (anti-helminthic agents), pimozide (anti-psychotic drug), omeprazole, pantoprazole (proton pump inhibitors) and many lead compounds are used in wide therapeutic areas.
Moreover, most of the drugs have 2nd and 5th position substitutions at the benzimidazole core. Substitutions at these two positions of the benzimidazole moiety were identified as essential for receptor activation and potency of receptor binding.28 On the other hand, [1,2,4]-oxadiazole is another privileged scaffold finding great importance in medicinal chemistry research due to its synthetic applications such as anticonvulsant,29 analgesic,30 anti-inflammatory,31 and antitumor32 applications. Also, [1,2,4]-oxadiazoles are reported for their selective inhibition of receptors such as tyrosinase inhibitors,33 muscarinic,34 histamine-H3 ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 35 and benzodiazepines.36 In addition, significant numbers of drugs and drug candidates in clinical development are halogenated. Insertion of halogen atoms in hit or lead compounds predominantly causes development in their steric effects by increasing the ability of bulky atoms to occupy the binding sites of molecular targets and also increases the polar hydrophobicity.37
35 and benzodiazepines.36 In addition, significant numbers of drugs and drug candidates in clinical development are halogenated. Insertion of halogen atoms in hit or lead compounds predominantly causes development in their steric effects by increasing the ability of bulky atoms to occupy the binding sites of molecular targets and also increases the polar hydrophobicity.37
Based on the biological profiles of the benzimidazoles and [1,2,4]-oxadiazole and also in continuation of our research work on synthesis of benzimidazoles by the ‘one pot’ nitro reductive cyclization method,38,39 we report the synthesis of a series of novel benzimidazoles with [1,2,4]-oxadiazole in a single molecular frame and their evaluation for antibacterial, antifungal and antimycobacterial activities (Fig. 1).
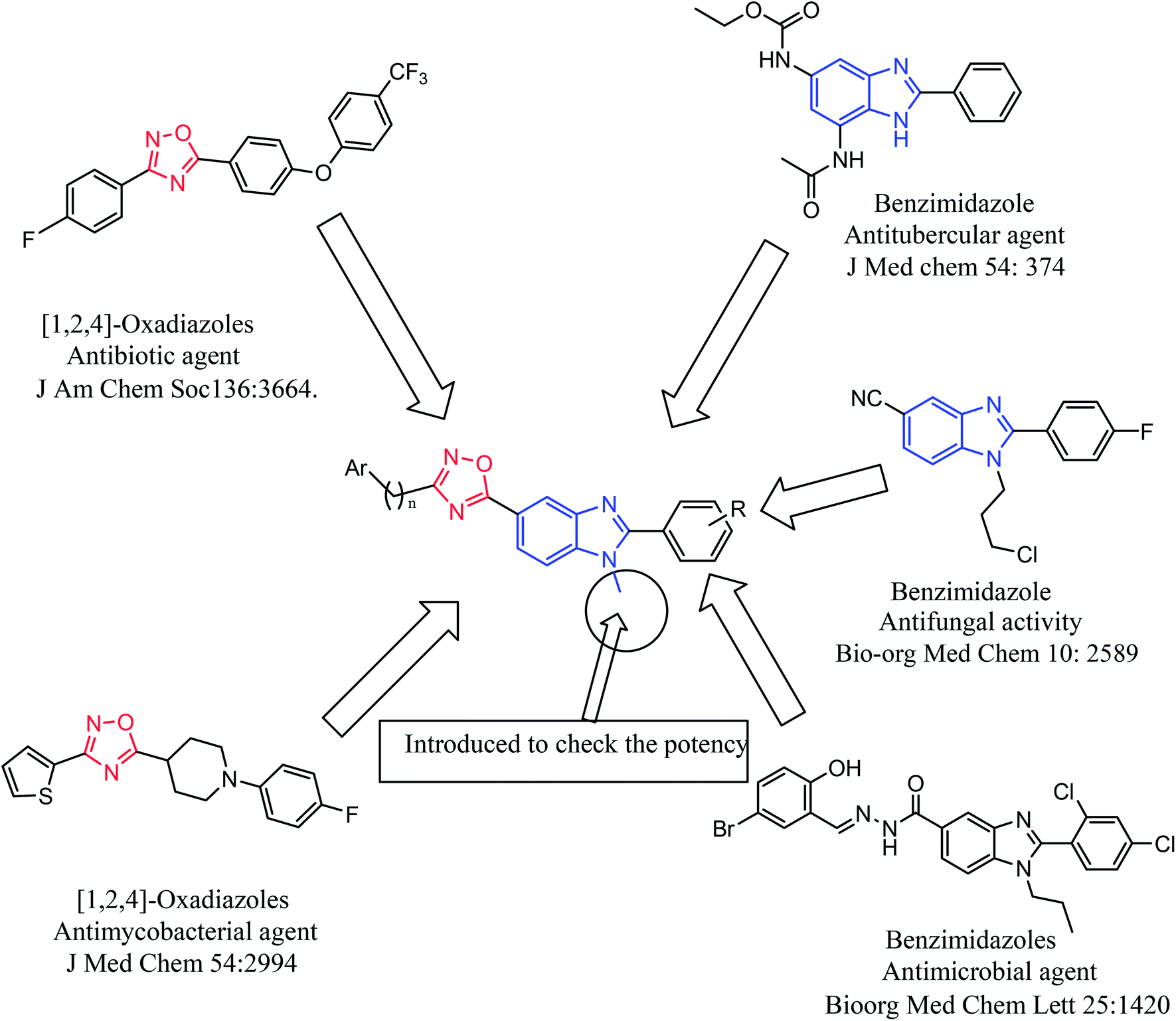 | ||
| Fig. 1 Design strategy for the generation of benzimidazoles with [1,2,4]-oxadiazoles scaffolds. | ||
2. Results and discussion
2.1. Chemistry
Twenty new compounds were synthesized by a series of reactions as depicted in Schemes 1–3. The intermediates N1-hydroxyamidines (2a–e) were synthesized by treating various aromatic/heterocyclic nitriles (1a–e) with hydroxylamine hydrochloride in ethanol using sodium bicarbonate. The intermediate ethyl 4-chloro-3-nitrobenzoate (4) was synthesized by esterification of commercially available 4-chloro-3-nitrobenzoic acid 3 according to a reported literature procedure.40 The compound ethyl 3-nitro-4-(methylamino)benzoate 5 was obtained by the nucleophilic displacement reaction of compound 4 with methylamine in the presence of triethylamine as a base in THF solvent. The core nucleus ethyl 2-aryl-1-methyl-1H-benzo[d]imidazole-5-carboxylate (6a–d) was prepared by ‘one-pot’ nitro reductive cyclization of 5 with various halo substituted aromatic aldehydes using sodium dithionite reagent in dimethyl sulphoxide medium at 90 °C. This ‘one pot’ reductive synthesis of 6a–d gave good yield (more than 90%) with high purity in very short reaction time as compared to conventional stepwise cyclization methods. Usually, in stepwise methods, the first step involves reduction of nitro group to an amine by hydrogenation or reduction reaction, followed by cyclization which requires strong acidic or oxidizing conditions. But this method involves several drawbacks, like harsh reaction conditions, use of expensive reagents, isolation of intermediates, tedious workup procedures, long reaction path and low yield. Hence, compared to stepwise methods, ‘one pot’ reductive cyclization is a simple, convenient, cheap and high yielding method which is much more superior to stepwise cyclization methods.38 The compound 2-aryl-1-methyl-1H-benzo[d]imidazole-5-carboxylic acid (7) was obtained by ester hydrolysis of ‘one pot’ intermediate 6a–d by using NaOH in ethanol medium. Finally, intermediates N1-hydroxyamidines (2a–e) undergo O-acylation with carboxylic acid group of intermediates 6a–d followed by intramolecular cyclodehydration in the presence of coupling agent, TBTU and base N-methylmorpholine in dimethylformamide medium affording 2-aryl-5-(3-aryl-[1,2,4]-oxadiazol-5-yl)-1-methyl-1H-benzo[d]imidazoles (8a–t). Physical properties and structural features of target compounds 8a–t are presented in Table 1.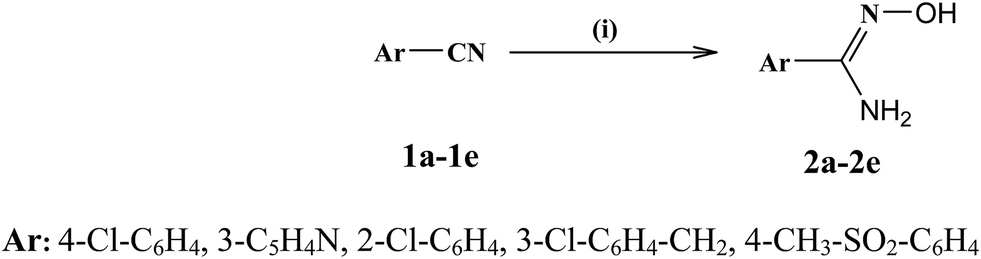 | ||
| Scheme 1 Synthesis of N1-hydroxyamidine 2a–e. Reagents and conditions: (i) hydroxylamine hydrochloride, sodium bicarbonate, EtOH, RT, 6 h. | ||
 | ||
| Scheme 2 Synthesis of core nucleus ethyl 2-aryl-1-methyl-1H-benzo[d]imidazole-5-carboxylate 6a–d. Reagents and conditions: (i) conc. H2SO4, EtOH, reflux, 6 h. (ii) Methylamine, triethylamine, THF, RT. (iii) Na2S2O4, DMSO, 90° C, 3 h. (iv) Sodium hydroxide, EtOH, reflux, 8 h. | ||
 | ||
| Scheme 3 Synthesis of target compounds, 2-aryl-5-(3-aryl-[1,2,4]-oxadiazol-5-yl)-1-methyl-1H-benzo[d]imidazoles 8a–t. Reagents and conditions: (i) TBTU, N-methyl morpholine, DMF, reflux, 8 h. | ||
|
|||||
|---|---|---|---|---|---|
| Compound | Ar | R | Mol. formula | Mol. weight | M. p. (°C) |
| 8a | 4-Chlorophenyl | 2-Chloro-6-fluoro | C22H13Cl2FN4O | 439.27 | 212–214 |
| 8b | Pyridine-3-yl | 2-Chloro-6-fluoro | C21H13ClFN5O | 405.81 | 222–224 |
| 8c | 2-Chlorophenyl | 2-Chloro-6-fluoro | C22H13Cl2FN4O | 439.27 | 202–204 |
| 8d | 3-Chlorobenzyl | 2-Chloro-6-fluoro | C23H15Cl2FN4O | 453.3 | 192–194 |
| 8e | 4-(Methylsulfonyl)phenyl | 2-Chloro-6-fluoro | C23H16ClFN4O3S | 482.91 | >225 |
| 8f | 4-Chlorophenyl | 2,4-Dichloro | C23H15Cl2FN4O | 455.72 | 200–202 |
| 8g | Pyridine-3-yl | 2,4-Dichloro | C21H13Cl2N5O | 422.27 | 188–190 |
| 8h | 2-Chlorophenyl | 2,4-Dichloro | C22H13Cl3N4O | 455.72 | 201–203 |
| 8i | 3-Chlorobenzyl | 2,4-Dichloro | C23H15Cl3N4O | 469.75 | 198–200 |
| 8j | 4-(Methylsulfonyl)phenyl | 2,4-Dichloro | C23H16Cl2N4O3S | 499.37 | 184–186 |
| 8k | 4-Chlorophenyl | 3-Chloro-2-fluoro | C22H13Cl2FN4O | 439.27 | >225 |
| 8l | Pyridine-3-yl | 3-Chloro-2-fluoro | C21H13ClFN5O | 405.81 | 186–189 |
| 8m | 2-Chlorophenyl | 3-Chloro-2-fluoro | C22H13Cl2FN4O | 439.27 | 190–192 |
| 8n | 3-Chlorobenzyl | 3-Chloro-2-fluoro | C23H15Cl2FN4O | 453.3 | >225 |
| 8o | 4-(Methylsulfonyl)phenyl | 3-Chloro-2-fluoro | C23H16ClFN4O3S | 482.91 | >225 |
| 8p | 4-Chlorophenyl | 2,3-Dichloro | C23H15Cl2FN4O | 455.72 | 194–196 |
| 8q | Pyridine-3-yl | 2,3-Dichloro | C21H13Cl2N5O | 422.27 | >225 |
| 8r | 2-Chlorophenyl | 2,3-Dichloro | C22H13Cl3N4O | 455.72 | 180–182 |
| 8s | 3-Chlorobenzyl | 2,3-Dichloro | C23H15Cl3N4O | 469.75 | 172–174 |
| 8t | 4-(Methylsulfonyl)phenyl | 2,3-Dichloro | C23H16Cl2N4O3S | 499.37 | >225 |

The newly synthesized compounds were characterized by FT (ATR) IR, 1H NMR, 13C NMR, ESI MS and elemental analyses. The formation of intermediates 2a–e from corresponding aromatic nitriles was confirmed by the appearance of characteristic stretching vibrations for NH and OH groups. For the compound 2e, NH and OH stretching vibrations were observed at 3342 and 3197 cm−1. The characteristic stretching vibrations for SO2 group were observed at 1382 cm−1 (asym) and 1123 cm−1 (sym). Also, in the 1H NMR spectrum, free NH2 signal appeared at δ 5.45 ppm and a distinct singlet for OH appeared at δ 8.93 ppm. Further, a singlet at δ 2.71 ppm indicated the presence of three protons of methylsulfonyl group. Two distinct doublets were observed at δ 7.47 and δ 7.60 ppm for four protons of methylsulfonyl phenyl ring, each integrating for two protons with coupling constants J = 8.4 Hz and 8.0 Hz respectively.
The formation of ‘one pot’ reductive cyclized core nucleus 6a–d was confirmed by the absence of characteristic NH stretching at 3350 cm−1 of the methylamine compound 5 of the previous step. Further, by considering prototype 6a, its 1H NMR spectrum showed three protons of 2-chloro-6-fluorophenyl ring as a triplet at δ 7.52 ppm (coupling constant J = 8.4 Hz) and a multiplet in the region δ 7.42–7.46 ppm which clearly indicated the cyclization. In addition, the spectrum displayed a doublet centered at δ 7.62 ppm, a doublet of doublet in the region of δ 7.71–7.81 ppm and a singlet at δ 8.34 ppm due to three aromatic protons of benzimidazole ring. The –CH3 and –CH2 protons of ester group appeared as a triplet at δ 1.36 ppm and a quartet in the region δ 4.33–4.39 ppm. In its spectrum, the disappearance of a broad –NH peak in the downfield region strongly confirmed the formation of compound 6a. Its formation was also evidenced by ESI MS spectral data which showed (M+ + H) peak at m/z value of 333.1. In the next step, the FT (ATR) IR spectrum of compound 7a showed the characteristic OH vibration at 3220 cm−1 which was absent in the spectrum of the corresponding previous ester 6a. The shifting of carbonyl stretching frequency to a lower value of 1693 cm−1 from 1735 cm−1 gave strong evidence for the conversion of ester group to acid. Further, its 1H NMR spectrum displayed a broad OH signal at δ 12.82 ppm and also the absence of signals due to ester protons in the shielded region strongly supported the conversion of ester 6a to acid 7a. In its ESI MS spectrum, the presence of a deprotonated molecular ion peak at m/z value of 303.0 further confirmed its formation.
Formation of new target compounds 8a–t was also characterized by spectral data and elemental analyses. In the FT (ATR) IR spectra, the absence of characteristic OH/NH vibrations strongly evidenced the formation of 8a–t, which was found in the spectra of intermediates 2a–e. Aromatic protons showed stretching vibrations around 3018–3080 cm−1. For the prototype compound 8a, C–F and C–Cl stretching bands were observed at 1165 cm−1 and 764 cm−1 respectively. Further, the 1H NMR spectrum of 8a showed a characteristic singlet for N–CH3 group at δ 3.67 ppm. Aromatic protons of 2-chloro-6-fluoro ring resonated as a multiplet in the region δ 7.42–7.46 ppm. The proton at C7 of benzimidazole appeared as a doublet at δ 7.34 ppm with coupling constant J = 7.6 Hz, C6 as a doublet of doublet in the region δ 8.17–8.19 ppm (J = 8.4 and 1.2 Hz) and C4 as a broad singlet at 8.65 ppm. In its13 C NMR spectrum, the signal due to carbonyl group was absent. N–CH3 carbon of benzimidazole gave a signal in the region δ 30–34 ppm, along with all aromatic carbons in the region δ 110–160 ppm. Oxadiazole ring carbons showed signals in the downfield region δ 162–176 ppm, which provide an additional support for the formation of the compound. Further, compound 8a displayed a protonated peak at m/z value of 439.2 along with isotopic peaks at m/z values of 441.2 and 443.2 in its ESI MS spectrum which is in accordance with its molecular formula weight and evidences the formation of the target molecule. The spectral and elemental analysis data of all target compounds are presented in the experimental section.
2.2. Biological screening
| Sample | Antibacterial activity (MIC in μg mL−1) | Antifungal activity (MIC in μg mL−1) | ||||
|---|---|---|---|---|---|---|
| Gram positive | Gram negative | |||||
| S. aureus | E. faecalis | E. coli | K. pneumoniae | C. albicans | A. fumigates | |
| a Bold font indicates a significant activity of compounds against the respective bacterial strains. | ||||||
| Control | — | — | — | — | — | — |
| 8a | 3.125 | 1.6 | 3.125 | 3.125 | 1.6 | 3.125 |
| 8b | 25.0 | 50.0 | 12.5 | 25.0 | 12.5 | 25.0 |
| 8c | 25.0 | 25.0 | 6.25 | 25.0 | 12.5 | 25.0 |
| 8d | 50.0 | 25.0 | 50.0 | 50.0 | 12.5 | 12.5 |
| 8e | 3.125 | 3.125 | 12.5 | 12.5 | 6.25 | 3.125 |
| 8f | 6.25 | 3.125 | 25.0 | 12.5 | 3.125 | 6.25 |
| 8g | 25.0 | 50.0 | 12.5 | 25.0 | 25 | 12.5 |
| 8h | 12.5 | 25.0 | 25.0 | 12.5 | 6.25 | 12.5 |
| 8i | 12.5 | 6.25 | 25.0 | 50.0 | 12.5 | 6.25 |
| 8j | 6.25 | 12.5 | 6.25 | 12.5 | 3.125 | 3.125 |
| 8k | 1.6 | 1.6 | 3.125 | 6.25 | 0.8 | 1.6 |
| 8l | 25.0 | 12.5 | 50.0 | 50.0 | 25 | 25 |
| 8m | 12.5 | 12.5 | 12.5 | 12.5 | 3.125 | 3.125 |
| 8n | 1.6 | 3.125 | 3.125 | 12.5 | 1.6 | 1.6 |
| 8o | 6.25 | 12.5 | 12.5 | 12.5 | 25 | 25.0 |
| 8p | 6.25 | 3.125 | 12.5 | 6.25 | 1.6 | 1.6 |
| 8q | 3.125 | 3.125 | 6.25 | 12.5 | 3.125 | 1.6 |
| 8r | 3.125 | 1.6 | 3.125 | 6.25 | 3.125 | 6.25 |
| 8s | 6.25 | 6.25 | 3.125 | 3.125 | 1.6 | 1.6 |
| 8t | 25.0 | 12.5 | 50.0 | 50.0 | 12.5 | 6.25 |
| Ciprofloxacin | 2.0 | 1.0 | 2.0 | 1.0 | — | — |
| Fluconazole | — | — | — | — | 16.0 | 8.0 |
| No. | Compound | MIC (μg mL−1) | HC50 (μg mL−1) | Selectivity ratio (HC50/MIC) |
|---|---|---|---|---|
| a ND = not determined. | ||||
| Control | — | — | — | |
| 1 | 8a | 3.125 | 226.72 | 72.55 |
| 2 | 8e | 25.0 | ND | — |
| 3 | 8f | 3.125 | ND | — |
| 4 | 8j | 25.0 | ND | — |
| 5 | 8k | 1.6 | 874.29 | 546.30 |
| 6 | 8n | 1.6 | 93.69 | 58.55 |
| 7 | 8p | 1.6 | 129.46 | 80.91 |
| 8 | 8q | 12.5 | ND | — |
| 9 | 8r | 1.6 | 369.05 | 230.63 |
| 10 | 8s | 3.125 | 280.84 | 89.86 |
| 11 | Isoniazid | 6.25 | ND | — |
| 12 | Pyrazinamide | 3.125 | ND | — |
| 13 | Streptomycin | 6.25 | 1016.34 | 162.56 |
| 14 | Ciprofloxacin | 3.125 | ND | — |
 | ||
| Fig. 2 Dose response curves of the cytotoxic effect of selected compounds on HEK 293 cell line. Values are the mean ± standard deviation of three independent experiments. | ||
| No. | Compound | IC50 value (μg mL−1) |
|---|---|---|
| 1 | 8k | >500 |
| 2 | 8n | >500 |
| 3 | 8p | >500 |
| 4 | 8r | 355.42 |
| 5 | Mitomycin C | <100 |
P) should be ≤5, and number of hydrogen bond donors should be ≤5 and number of hydrogen bond acceptors ≤10. Along with this rule, the number of rotatable bonds in a molecule should be ≤10 and topological polar surface area (TPSA) should be ≤140 Å2 for an effective orally bioavailable compound. TPSA is the sum of surfaces of polar atoms such as oxygen, nitrogen and attached hydrogen and the number of rotatable bonds is a measure of conformational flexibility of the molecule which in turn decides the binding of receptors. The percentage absorption was calculated by the equation %ABS = [109 − (0.345)TPSA].In our present study, the physicochemical descriptors of the potent compounds 8a, 8f, 8k, 8n, 8p, 8r and 8s were assessed to determine the ADME parameters by using the Molinspiration online property calculation toolkit and the results are summarized in Table 5. From the table we can observe that all the compounds showed excellent passive oral absorption with percentage absorption >90%. All the target compounds satisfy all the parameters except the logP value, which is slightly higher than the limiting value for all the compounds. But, violation of any one of the rules does not affect the bioavailability of the molecule and drug-likeliness,49 if the percentage absorption is good. TPSA was found to be 56.75 Å2 for all the compounds and numbers of rotatable bonds were less than 10 for all the compounds which decide molecular flexibility.
| Compound | MV | MW | milogP |
nON | nOHNH | nROTB | %ABS | TPSA (Å2) | nVioltn |
|---|---|---|---|---|---|---|---|---|---|
| a MV: molecular volume; MW: molecular weight; milogP: logarithm of partition coefficient between n-octanol and water; nON: number of hydrogen bond donors; nOHNH: number of hydrogen bond donors; nROTB: number of rotatable bonds, TPSA: topological polar surface area; %ABS: percentage absorption; nVioltn: number of violations. |
|||||||||
| 8a | 345.29 | 439.28 | 6.70 | 5 | 0 | 3 | 89.42 | 56.75 | 1 |
| 8f | 353.89 | 455.73 | 7.34 | 5 | 0 | 3 | 89.42 | 56.75 | 1 |
| 8k | 345.29 | 439.28 | 6.80 | 5 | 0 | 3 | 89.42 | 56.75 | 1 |
| 8n | 362.09 | 453.30 | 6.73 | 5 | 0 | 4 | 89.42 | 56.75 | 1 |
| 8p | 353.89 | 455.73 | 7.32 | 5 | 0 | 3 | 89.42 | 56.75 | 1 |
| 8r | 353.89 | 455.73 | 7.27 | 5 | 0 | 3 | 89.42 | 56.75 | 1 |
| 8s | 347.08 | 469.75 | 7.30 | 5 | 0 | 4 | 89.42 | 56.75 | 1 |
3. Conclusion
In conclusion, this work is focused on the development of novel potent compounds based on the benzimidazole–oxadiazole compact system. A series of twenty novel compounds of benzimidazoles with [1,2,4]-oxadiazoles were designed and efficiently synthesized with good yield by employing rapid ‘one pot’ nitro reductive cyclization. Compounds 8a and 8k emerged as promising antimicrobial agents against all the bacterial and fungal pathogens tested and are found to be superior to ciprofloxacin and fluconazole with good ADME parameters. Compounds 8k and 8n with 3-chloro-2-fluoro aryl ring at the 2nd position of benzimidazole and 4-chlorophenyl (8k) and 3-chorobenzyl (8n) substitution at oxadiazole ring emerged as excellent anti-tubercular agents with twofold more activity than pyrazinamide and ciprofloxacin, and four times more than streptomycin and isoniazid. Also, compounds 8p and 8r with 2,3-dichlorophenyl substitutions at benzimidazole end and 4-chloro (8p) and 2-chlorophenyl (8r) substitutions at oxadiazole phenyl ring are found to be around two times more active than pyrazinamide and ciprofloxacin, and fourfold more than streptomycin and isoniazid against M. tuberculosis M37Rv, and which emerged as potent anti-tuberculosis agents along with good drug-like properties. The preliminary toxicity testing of potent compounds 8a, 8k, 8n, 8p, 8r and 8s against human erythrocytes showed less toxicity and good selectivity index. Compound 8k showed the highest selectivity index of 546-fold. Further investigation of cytotoxicity of potent compounds against human embryonic kidney cell line HEK 293 revealed the nontoxicity of these compounds. Structure–activity relationships revealed that the presence of dihalo substitution at the 2nd position aryl ring of benzimidazole moiety as well as halogen substitution at the phenyl ring of [1,2,4]-oxadiazole are crucial for pronounced activity and significantly enhanced antibacterial, antifungal and antimycobacterial activities. Further detailed structural modifications at 1st, 2nd and 5th positions of benzimidazole moiety with different heterocyclic rings are underway to get better analogs with improved efficacy.4. Experimental section
4.1. Materials and methods
4.1.1.1. 4-Chloro-N′-hydroxybenzamidine (2a). White crystalline solid; FT IR (ATR, νmax, cm−1): 3348 (N–H), 3207 (OH), 3030 (Ar–H), 2979, 2928 (C–H), 1652 (C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N); 1H NMR (400 MHz, DMSO-d6, δ ppm): 8.98 (s, OH), 7.78 (d, 2H, p-Cl phenyl ring, J = 8.8 Hz), 7.50 (d, 2H, p-Cl phenyl ring, J = 8.4 Hz), 5.45 (s, 2H, NH2); ESI-MS: 171.1 (M+ + H), 173.1 [(M + H) + 2]+; anal. calcd for C7H7ClN2O: C, 49.28; H, 4.14; N, 16.42. Found: C, 49.27; H, 4.15; N, 16.41.
N); 1H NMR (400 MHz, DMSO-d6, δ ppm): 8.98 (s, OH), 7.78 (d, 2H, p-Cl phenyl ring, J = 8.8 Hz), 7.50 (d, 2H, p-Cl phenyl ring, J = 8.4 Hz), 5.45 (s, 2H, NH2); ESI-MS: 171.1 (M+ + H), 173.1 [(M + H) + 2]+; anal. calcd for C7H7ClN2O: C, 49.28; H, 4.14; N, 16.42. Found: C, 49.27; H, 4.15; N, 16.41.
4.1.1.2. N′-Hydroxynicotinamidine (2b). Off white solid; FT IR (ATR, νmax, cm−1): 3350 (N–H), 3200 (OH), 3020 (Ar–H), 2989, 2933 (C–H), 1650 (C
N); 1H NMR (400 MHz, DMSO-d6, δ ppm): 5.49 (s, 2H, NH2), 7.58 (d, 1H, Ar–H, J = 8.4 Hz), 8.03 (s, 1H, Ar–H), 8.16–8.20 (m, 2H, Ar–H), 8.98 (s, OH); ESI-MS: 138.14 (M+ + H); anal. calcd for C6H7N3O: C, 52.55; H, 5.14; N, 30.64. Found: C, 52.54; H, 5.15; N, 30.65.
4.1.1.3. 2-Chloro-N′-hydroxybenzamidine (2c). White crystalline solid; FT IR (ATR, νmax, cm−1): 3360 (N–H), 3220 (OH), 3025 (Ar–H), 2980, 2930 (C–H), 1649 (C
N); 1H NMR (400 MHz, DMSO-d6, δ ppm): 5.49 (s, 2H, NH2), 7.26–7.37 (m, 2H, Ar–H), 7.42 (m, 1H, Ar–H), 7.52 (dd, 1H, Ar–H, J = 7.5 and 1.4 Hz), 8.98 (s, OH); ESI-MS: 171.6 (M + H)+, 173.3 [(M + H) + 2]+; anal. calcd for C7H7ClN2O: C, 49.28; H, 4.14; N, 16.42. Found: C, 49.27; H, 4.15; N, 16.43.
4.1.1.4. 3-Chlorophenyl-N′-hydroxyacetamidine (2d). White crystalline solid; FT IR (ATR, νmax, cm−1): 3366 (N–H), 3204 (OH), 3015 (Ar–H), 2985, 2937 (C–H), 1649 (C
N); 1H NMR (400 MHz, DMSO-d6, δ ppm): 3.32 (S, 2H, benzyl–CH2), 7.93 (d, 1H, Ar–H, J = 8.8 Hz), 8.08 (t, 1H, Ar–H, J = 8.4 Hz), 8.26 (d, 1H, Ar–H, J = 8.4 Hz), 8.48 (s, 1H, Ar–H); ESI-MS: 185.06 (M+ + H), 187.4 [(M + H) + 2]+; anal. calcd for C8H9ClN2O: C, 52.04; H, 4.91; N, 15.17. Found: C, 52.05; H, 4.90; N, 15.18.
4.1.1.5. N′-Hydroxy-4-(methylsulfonyl)benzamidine (2e). Light brown crystals; FT IR (ATR, νmax, cm−1): 3414 (OH), 3263 (NH), 3081 (Ar–H), 2968 (C–H), 1653 (C
N), 1379 (asym SO2), 1083 (sym SO2); 1H NMR (400 MHz, DMSO-d6, δ ppm): 2.71 (s, 2H, –CH3), 5.45 (s, 2H, NH2), 7.47 (d, 2H, Ar–H, J = 8.4 Hz), 7.60 (d, 2H, Ar–H, J = 8 Hz), 8.93 (s, OH); ESI-MS: 214.5 (M+ + H), 215.1 [(M + H) + 1]+; anal. calcd for C8H10N2O3S: C, 44.85; H, 4.70; N, 13.08; S, 14.97. Found: C, 44.86; H, 4.71; N, 13.09; S, 14.98.
:1).Yellow solid; FT IR (ATR, νmax, cm−1): 3097 (Ar–H), 2976, 2941 (C–H), 1535 (NO2 asym), 1354 (NO2 sym); 1H NMR (400 MHz, DMSO-d6, δ ppm): 1.34 (t, 3H, –CH3, J = 7.2 Hz), 4.34–4.39 (q, 2H, ester CH2), 7.94 (d, 1H, Ar–H), 8.18–8.21 (dd, 1H, Ar–H, J = 8.4 Hz and J = 2 Hz), 8.51 (s, 1H, Ar–H); ESI-MS: 232.0 [(M + H) + 1]+, 234.1 [(M + H) + 3]+; anal. calcd for C9H8ClNO4: C, 47.08; H, 3.51; N, 15.44. Found: C, 47.07; H, 3.50; N, 15.43.
Light yellow solid; FT IR (ATR, νmax, cm−1): 3369 (N–H), 3037 (Ar–H), 2926, 2883 (C–H), 1535 (NO2 asym), 1354 (NO2 sym); 1H NMR (400 MHz, DMSO-d6, δ ppm): 1.36 (t, 3H, –CH3, J = 7.2 Hz), 3.01 (s, 3H, –CH3), 4.34–4.39 (q, 2H, ester CH2), 6.75 (d, 1H, Ar–H, J = 8.8 Hz), 7.59–7.72 (m, 1H, Ar–H), 8.11 (s, 1H, Ar–H), 12.41 (s, NH); ESI-MS: 224.5 (M+ − H); anal. calcd for C10H12N2O4: C, 53.57; H, 5.39; N, 12.49. Found: C, 53.58; H, 5.39; N, 12.48.
:hexane 3:7, v/v), it was poured into crushed ice. The solid separated was filtered off, washed with water, and dried. Collected product was recrystallized from dimethylformamide.
4.1.4.1. Ethyl 2-(2-chloro-6-flourophenyl)-1-methyl-1H-benzo[d]imidazole-5-carboxylate (6a). White powder; FT IR (ATR, νmax, cm−1): 3061 (Ar–H), 2972, 2926 (C–H), 1699 (C
O), 1620 (CN), 1284 (C–F), 896 (C–Cl); 1H NMR (400 MHz, DMSO-d6, δ ppm): 1.36 (t, 3H, J = 7.2 Hz, –CH3), 3.70 (s, 3H, –CH3), 4.33–4.39 (q, 2H, –CH2–CH3, J = 14.4 and 6.8 Hz), 7.52 (t, 1H, Ar–H, J = 8.4 Hz), 7.62 (d, 1H, Ar–H, J = 7.2 Hz), 7.71–7.77 (m, 1H, Ar–H), 7.80 (d, 1H, Ar–H, J = 8.4 Hz), 7.98–8.01 (dd, 1H, Ar–H, J = 8.8 and 1.6 Hz), 8.34 (s, 1H, Ar–H); ESI-MS (m/z): 333.1 (M+ + H), 335.1 [(M + H) + 2]+; anal. calcd for C17H14ClFN2O2: C, 61.36; H, 4.24; N, 8.42. Found: C, 61.32; H, 4.25; N, 8.46.
4.1.4.2. Ethyl 2-(2,4-dichlorophenyl)-1-methyl-1H-benzo[d]imidazole-5-carboxylate (6b). White amorphous solid; FT IR (ATR, νmax, cm−1): 3040 (Ar–H), 2983, 2932 (C–H), 1692 (C
O), 1614 (CN), 1538 (CC), 890 (C–Cl); 1H NMR (400 MHz, DMSO-d6, δ ppm): 1.38 (t, 3H, J = 7.2 Hz, –CH3), 3.73 (s, 3H, –CH3), 4.34–4.40 (q, 2H, –CH2–CH3, J = 14.4 and 6.8 Hz), 7.58 (s, 1H, Ar–H), 7.66 (d, 1H, Ar–H, J = 7.2 Hz), 7.98–8.01 (dd, 1H, Ar–H, J = 8.8 and 1.6 Hz), 8.09 (d, 2H, Ar–H, J = 8.8 Hz), 8.37 (s, 1H, Ar–H); ESI-MS (m/z): 349.1 (M+ + H), 351.6 [(M + H) + 2]+, 353.4 [(M + H) + 4]+; anal. calcd for C17H14Cl2N2O2: C, 58.47; H, 4.04; N, 8.02. Found: C, 58.48; H, 4.07; N, 8.02.
4.1.4.3. Ethyl 2-(3-chloro-2-fluorophenyl)-1-methyl-1H-benzo[d]imidazole-5-carboxylate (6c). White amorphous solid; FT IR (ATR, νmax, cm−1): 3045 (Ar–H), 2981, 2931 (C–H), 1699 (C
O), 1609 (CN), 1539 (CC), 1272 (C–F), 890 (C–Cl); 1H NMR (400 MHz, DMSO-d6, δ ppm): 1.36 (t, 3H, 7.2 Hz, –CH3), 3.73 (s, 3H, –CH3), 4.32–4.39 (q, 2H, –CH2–CH3, J = 14.4 and 6.8 Hz), 7.42–7.47 (m, 3H, 3-Cl-2-F phenyl ring), 7.69 (d, 1H, Ar–H, J = 7.2 Hz), 7.99–8.02 (dd, 1H, Ar–H, J = 8.8 and 1.6 Hz), 8.38 (s, 1H, Ar–H); ESI-MS (m/z): 333.1 (M+ + H), 335.7 [(M + H) + 2]+; anal. calcd for C17H14ClFN2O2: C, 61.36; H, 4.24; N, 8.42. Found: C, 61.38; H, 4.22; N, 8.43.
4.1.4.4. Ethyl 2-(2,3-dichlorophenyl)-1-methyl-1H-benzo[d]imidazole-5-carboxylate (6d). White crystalline solid; FT IR (ATR, νmax, cm−1): 3049 (Ar–H), 2990, 2939 (C–H), 1700 (C
O), 1623 (CN), 1539 (CC), 889 (C–Cl); 1H NMR (400 MHz, DMSO-d6, δ ppm): 1.38 (t, 3H, 7.2 Hz, –CH3), 3.75 (s, 3H, –CH3), 4.31–4.39 (q, 2H, –CH2–CH3, J = 14.4 and 6.8 Hz), 7.60 (d, 1H, Ar–H, J = 7.2 Hz), 7.68 (d, 1H, Ar–H, J = 8.4 Hz), 7.83 (t, 1H, Ar–H, J = 8.4 Hz), 7.99–8.02 (dd, 1H, Ar–H, J = 8.8 and 1.6 Hz), 8.09 (d, 1H, Ar–H, J = 8.8 Hz), 8.38 (s, 1H, Ar–H); ESI-MS (m/z): 350.1 (M+ + H), 352.6 [(M + H) + 2]+, 354.2 [(M + H) + 4]+; anal. calcd for C17H14Cl2N2O2: C, 58.47; H, 4.04; N, 8.02. Found: C, 58.45; H, 4.05; N, 8.04.
4.1.5.1. 2-(2-Chloro-6-fluorophenyl)-1-methyl-1H-benzo[d]imidazole-5-carboxylic acid (7a). Creamy white powder; FT IR (ATR, νmax, cm−1): 3196 (OH), 3030 (Ar–H), 2970, 2924 (C–H), 1689 (C
O), 1618 (CN), 1566 (CC), 1033 (C–F), 752 (C–Cl); 1H NMR (400 MHz, DMSO-d6, δ ppm): 3.69 (s, 1H, –CH3), 7.52 (t, 1H, Ar–H, J = 8.8 Hz), 7.61 (d, 1H, Ar–H, J = 8 Hz), 7.71–7.78 (m, 2H, Ar–H), 7.98 (d, 1H, Ar–H, J = 8.4 Hz), 8.30 (s, 1H, Ar–H), 12.82 (bs, OH, 1H); ESI-MS (m/z): 305.0 (M+ + H), 307.1 [(M + H) + 2]+. Anal. calcd for C15H10ClFN2O2: C, 59.13; H, 3.31; N, 9.19. Found: C, 59.16; H, 3.32; N, 9.22.
4.1.5.2. 2-(2,4-Dichlorophenyl)-1-methyl-1H-benzo[d]imidazole-5-carboxylic acid (7b). White amorphous solid; FT IR (ATR, νmax, cm−1): 3240 (OH), 2984, 2939 (C–H), 1684 (C
O), 1616 (CN), 1528 (CC), 895 (C–Cl); 1H NMR (400 MHz, DMSO-d6, d ppm): 3.73 (s, 3H, –CH3), 7.59 (s, 1H, Ar–H), 7.66 (d, 1H, Ar–H, J = 7.2 Hz), 7.99–8.02 (dd, 1H, Ar–H, J = 8.8 and 1.6 Hz), 8.09 (d, 2H, Ar–H, J = 8.8 Hz), 8.37 (s, 1H, Ar–H), 12.80 (bs, OH, 1H); ESI-MS (m/z): 320.1 (M+ − H), 322.4 (M + H)+; anal. calcd for C15H10Cl2N2O2: C, 56.10; H, 3.14; N, 8.72. Found: C, 56.08; H, 3.17; N, 8.75.
4.1.5.3. 2-(3-Chloro-2-fluorophenyl)-1-methyl-1H-benzo[d]imidazole-5-carboxylic acid (7c). Light cream solid; FT IR (ATR, νmax, cm−1): 3194 (OH), 2981, 2931 (C–H), 1689 (C
O), 1599 (CN), 1539 (CC), 1279 (C–F), 892 (C–Cl); 1H NMR (400 MHz, DMSO-d6, δ ppm): 3.73 (s, 3H, –CH3), 7.43–7.48 (m, 3H, 3-Cl-2-F phenyl ring), 7.69 (d, 1H, Ar–H, J = 7.2 Hz), 7.99–8.02 (dd, 1H, Ar–H, J = 8.8 and 1.6 Hz), 8.38 (s, 1H, Ar–H), 12.73 (bs, OH, 1H); ESI-MS (m/z): 305.7 (M+ + H), 307.2 [(M + H) + 2]+; anal. calcd for C15H10ClFN2O2: C, 59.13; H, 3.31; N, 9.19. Found: C, 59.10; H, 3.34; N, 9.18.
4.1.5.4. 2-(2,3-Dichlorophenyl)-1-methyl-1H-benzo[d]imidazole-5-carboxylic acid (7d). White amorphous sold; FT IR (ATR, νmax, cm−1): 3149 (OH), 2990, 2939 (C–H), 1690 (C
O), 1603 (CN), 1549 (CC), 886 (C–Cl); 1H NMR (400 MHz, DMSO-d6, δ ppm): 3.75 (s, 3H, –CH3), 7.60 (d, 1H, Ar–H, J = 7.2 Hz), 7.68 (d, 1H, Ar–H, J = 8.4 Hz), 7.83 (t, 1H, Ar–H, J = 8.4 Hz), 7.99–8.02 (dd, 1H, Ar–H, J = 8.8 and 1.6 Hz), 8.09 (d, 1H, Ar–H, J = 8.8 Hz), 8.38 (s, 1H, Ar–H), 12.43 (bs, OH, 1H); ESI-MS (m/z): 322.1 (M+ + H), 324.3 [(M + H) + 2]+, 326.3 [(M + H) + 4]+; anal. calcd for C15H10Cl2N2O2: C, 56.10; H, 3.14; N, 8.72. Found: C, 56.14; H, 3.15; N, 8.74.
4.1.6.1. 2-(2-Chloro-6-fluorophenyl)-5-(3-(4-chlorophenyl)-[1,2,4]-oxadiazol-5-yl)-1-methyl-1H-benzo[d]imidazole (8a). Pale yellow solid; FT IR (ATR, νmax, cm−1): 3018 (Ar–H), 2976, 2945 (C–H), 1618 (C
N), 1596 (CC), 1165 (C–F), 754 (C–Cl); 1H NMR (400 MHz, DMSO, δ ppm): 3.67 (s, 3H, –CH3), 7.34 (d, 2H, Ar–H, J = 7.6 Hz), 7.42–7.46 (m, 3H, 2-Cl-6-F phenyl ring), 7.52 (d, 2H, p-Cl phenyl ring, J = 8.4 Hz), 8.08 (d, 2H, p-Cl phenyl ring, J = 8.8 Hz), 8.17–8.19 (dd, 1H, J = 8.4 Hz and 1.2 Hz), 8.65 (s, 1H, benzimidazole-H); 13C NMR (125 MHz, DMSO, δ ppm): 30.72 (–CH3), 110.56, 114.60, 118.63, 121.17, 123.31, 125.78, 128.85, 132.63, 132.70, 137.18, 138.44, 143.06, 160.43, 162.44 (–CN), 168.13 (–CN), 176.53 (–CN); ESI-MS: 439.2 (M+ + H), 441.2 [(M + H) + 2]+, 443.2 [(M + H) + 4]+; anal. calcd for C22H13Cl2FN4O: C, 60.15; H, 2.98; N, 12.75. Found: C, 60.18; H, 2.99; N, 12.79.
4.1.6.2. 2-(2-Chloro-6-fluorophenyl)-5-(3-(pyridin-3-yl)-[1,2,4]-oxadiazol-5-yl)-1-methyl-1H-benzo[d]imidazole (8b). Yellow solid; FT IR (ATR, νmax, cm−1): 3028 (Ar–H), 2996, 2935 (C–H), 1608 (C
N), 1596 (CC), 1145 (C–F), 784 (C–Cl); 1H NMR (400 MHz, DMSO, δ ppm): 3.69 (s, 3H, –CH3), 7.43–7.47 (m, 3H, 2-Cl-6-F phenyl ring), 7.54 (d, 1H, Ar–H, J = 8.4 Hz), 7.58 (d, 1H, Ar–H, J = 8.4 Hz), 8.08 (s, 1H, Ar–H), 8.16–8.20 (m, 2H, Ar–H), 8.24–8.27 (dd, 1H, benzimidazole-H, J = 8.4 Hz and 1.2 Hz), 8.69 (s, 1H, benzimidazole-H); 13C NMR (125 MHz, DMSO, δ ppm): 31.72 (–CH3), 111.50, 114.65, 119.64, 120.17, 122.61, 124.88, 129.87, 131.64, 132.75, 136.18, 139.44, 144.06, 148.37, 159.43, 161.47 (–CN), 169.13 (–CN), 177.53 (–CN); ESI-MS: 407.3 (M+ + H), 409.3 [(M + H) + 2]+; anal. calcd for C21H13ClFN5O: C, 62.15; H, 3.23; N, 17.26. Found: C, 62.16; H, 3.26; N, 17.28.
4.1.6.3. 2-(2-Chloro-6-fluorophenyl)-5-(3-(2-chlorophenyl)-[1,2,4]-oxadiazol-5-yl)-1-methyl-1H-benzo[d]imidazole (8c). White solid; FT IR (ATR, νmax, cm−1): 3054 (Ar–H), 2986, 2935 (C–H), 1618 (C
N), 1586 (CC), 1148 (C–F), 786 (C–Cl); 1H NMR (400 MHz, DMSO, δ ppm): 3.64 (s, 3H, –CH3), 7.34–7.38 (m, 3H, Ar–H, J = 8 Hz), 7.49 (d, 1H, Ar–H, J = 8.4 Hz), 7.53–7.56 (m, 4H, Ar–H), 8.20–8.22 (dd, 1H, benzimidazole-H, J = 8.8 Hz and 1.6 Hz), 8.60 (s, 1H, benzimidazole-H); 13C NMR (125 MHz, DMSO, δ ppm): 32.32 (–CH3), 113.55, 115.65, 120.64, 122.17, 124.65, 126.88, 127.33, 129.87, 132.64, 133.75, 136.58, 138.49, 144.06, 157.43, 160.47 (–CN), 166.13 (–CN), 177.53 (–CN); ESI-MS: 440.2 (M+ + H), 442.4 [(M + H) + 2]+, 444.7 [(M + H) + 4]+; anal. calcd for C22H13Cl2FN4O: C, 60.15; H, 2.98; N, 12.75. Found: C, 60.17; H, 2.96; N, 12.78.
4.1.6.4. 2-(2-Chloro-6-fluorophenyl)-5-(3-(3-chlorobenzyl)-[1,2,4]-oxadiazol-5-yl)-1-methyl-1H-benzo[d]imidazole (8d). White amorphous solid; FT IR (ATR, νmax, cm−1): 3069 (Ar–H), 2976, 2929 (C–H), 1602 (C
N), 1576 (CC), 1188 (C–F), 796 (C–Cl); 1H NMR (400 MHz, DMSO, δ ppm): 3.30 (s, 2H, benzyl-CH2), 3.71 (s, 3H, imidazole-CH3), 7.47–7.54 (m, 3H, 2-Cl-6-F phenyl ring-H), 7.59 (d, 1H, Ar–H, J = 8 Hz), 7.70–7.73 (m, 1H, Ar–H, benzimidazole-H), 7.76 (d, 1H, Ar–H, J = 8.4 Hz, benzimidazole-H), 7.90 (d, 1H, Ar–H, J = 8.8 Hz), 8.08 (t, 1H, Ar–H, J = 8.4 Hz), 8.43 (s, 1H, Ar–H), 8.57 (s, 1H, benzimidazole-H); 13C NMR (125 MHz, DMSO, δ ppm): 30.99 (–CH3), 36.46 (CH2), 110.79, 121.59, 124.24, 125.97, 126.64, 127.47, 127.82, 128.56, 128.52, 130.16, 146.92, 157.59, 163.92 (–CN), 169.53 (–CN), 175.86 (–CN); ESI-MS: 453.2 (M+ + H), 455.2 [(M + H) + 2]+, 457.2 [(M + H) + 4]+; anal. calcd for C23H15Cl2FN4O: C, 60.94; H, 3.34; N, 12.36. Found: C, 60.95; H, 3.35; N, 12.38.
4.1.6.5. 2-(2-Chloro-6-fluorophenyl)-5-(3-(4-(methylsulfonyl)phenyl)-[1,2,4]-oxadiazol-5-yl)-1-methyl-1H-benzo[d]imidazole (8e). White amorphous solid; FT IR (ATR, νmax, cm−1): 3099 (Ar–H), 2996, 2939 (C–H), 1613 (C
N), 1571 (CC), 1388 (asym SO2), 1185 (C–F), 1129 (sym SO2), 798 (C–Cl); 1H NMR (400 MHz, DMSO, δ ppm): 3.24 (s, 3H, CH3), 3.63 (s, 3H, CH3), 7.66–7.75 (m, 5H, Ar–H), 7.96 (d, 2H, Ar–H, J = 8.8 Hz), 8.12 (d, 2H, Ar–H, J = 8.4 Hz), 8.57 (s, 1H, Ar–H); 13C NMR (125 MHz, DMSO, δ ppm): 31.72 (–CH3), 111.56, 116.60, 119.63, 120.17, 126.73, 129.75, 133.63, 134.70, 136.62, 137.11, 139.49, 148.06, 161.43, 163.44 (–CN), 169.13 (–CN), 176.53 (–CN); ESI-MS: 483.2 (M+ + H), 484.2 [(M + H) + 2]+; anal. calcd for C23H16ClFN4O3S: C, 57.20; H, 3.34; N, 11.60; S, 6.64. Found: C, 57.21; H, 3.37; N, 11.63; S, 6.63.
4.1.6.6. 2-(2,4-Dichlorophenyl)-5-(3-(4-chlorophenyl)-[1,2,4]-oxadiazol-5-yl)-1-methyl-1H-benzo[d]imidazole (8f). Light yellow solid; FT IR (ATR, νmax, cm−1): 3077 (Ar–H), 2992, 2930 (C–H), 1623 (C
N), 1571 (CC), 778 (C–Cl); 1H NMR (400 MHz, DMSO, δ ppm): 3.68 (s, 3H, –CH3), 7.39 (d, 1H, benzimidazole-H, J = 8 Hz), 7.43 (d, 2H, 4-chlorophenyl, J = 8.4 Hz), 7.50 (d, 2H, 4-chlorophenyl, J = 8.4 Hz), 7.53 (s, 1H, 2,4-dichlorophenyl), 8.08 (d, 2H, 2,4-dichlorophenyl, J = 8.8 Hz), 8.16–8.19 (dd, 1H, benzimidazole-H, J = 8.4 Hz and 1.6 Hz), 8.62 (s, 1H, benzimidazole-H); 13C NMR (125 MHz, DMSO, δ ppm): 31.20 (–CH3), 111.55, 119.75, 121.97, 123.36, 125.62, 127.74, 128.86, 129.15, 129.88, 133.18, 137.22, 152.60 (–CN), 168.16 (–CN), 176.51 (–CN); ESI-MS: 455.1 (M+ + H), 457.1 [(M + H) + 2]+, 459.1 [(M + H) + 4]+, 461.0 [(M + H) + 6]+; anal. calcd for C23H15Cl2FN4O: C, 57.98; H, 2.88; N, 12.29. Found: C, 57.96; H, 2.86; N, 12.30.
4.1.6.7. 2-(2,4-Dichlorophenyl)-5-(3-(pyridin-3-yl)-[1,2,4]-oxadiazol-5-yl)-1-methyl-1H-benzo[d]imidazole (8g). Light brown solid; FT IR (ATR, νmax, cm−1): 3057 (Ar–H), 2994, 2932 (C–H), 1629 (C
N), 1576 (CC), 778 (C–Cl); 1H NMR (400 MHz, DMSO, δ ppm): 3.68 (s, 3H, –CH3), 7.40 (d, 2H, Ar–H, J = 8.4 Hz), 7.56 (s, 1H, Ar–H), 7.59 (d, 1H, Ar–H, J = 8 Hz), 7.63 (d, 1H, Ar–H, J = 7.6 Hz), 8.08–8.12 (m, 3H, Ar–H), 8.17–8.20 (dd, 1H, benzimidazole-H, J = 8.4 Hz and 1.2 Hz), 8.64 (s, 1H, benzimidazole-H); 13C NMR (125 MHz, DMSO, δ ppm): 30.20 (–CH3), 109.55, 116.57, 118.24, 121.98, 122.36, 124.62, 125.41, 127.74, 128.36, 129.45, 129.88, 130.31, 133.18, 137.22, 138.20, 154.64 (–CN), 169.16 (–CN), 175.59 (–CN); ESI-MS: 455.1 (M+ + H), 457.5 [(M + H) + 2]+; anal. calcd for C21H13Cl2N5O: C, 59.73; H, 3.10; N, 16.79. Found: C, 59.75; H, 3.11; N, 16.82.
4.1.6.8. 2-(2,4-Dichlorophenyl)-5-(3-(2-chlorophenyl)-[1,2,4]-oxadiazol-5-yl)-1-methyl-1H-benzo[d]imidazole (8h). White amorphous solid; FT IR (ATR, νmax, cm−1): 3057 (Ar–H), 2994, 2932 (C–H), 1629 (C
N), 1576 (CC), 778 (C–Cl); 1H NMR (400 MHz, DMSO, δ ppm): 3.64 (s, 3H, –CH3), 7.34 (d, 2H, Ar–H, J = 8.4 Hz), 7.52–7.55 (m, 4H, Ar–H), 7.77 (d, 1H, benzimidazole-H, J = 8 Hz), 7.87 (s, 1H, Ar–H), 8.22–8.25 (dd, 1H, J = 8.4 Hz and 1.2 Hz), 8.65 (s, 1H, benzimidazole-H); 13C NMR (125 MHz, DMSO, δ ppm): 30.29 (–CH3), 110.59, 118.45, 120.94, 124.36, 125.82, 127.76, 128.80, 129.85, 129.99, 133.18, 135.32, 136.22, 137.46, 140.39, 142.36, 153.60 (–CN), 166.76 (–CN), 174.71 (–CN); ESI-MS: 455.1 (M+ + H), 457.1 [(M + H) + 2]+, 459.5 [(M + H) + 4]+, 461.0 [(M + H) + 6]+; anal. calcd for C22H13Cl3N4O: C, 57.98; H, 2.88; N, 12.29. Found: C, 57.99; H, 2.89; N, 12.30.
4.1.6.9. 2-(2,4-Dichlorophenyl)-5-(3-(3-chlorobenzyl)-[1,2,4]-oxadiazol-5-yl)-1-methyl-1H-benzo[d]imidazole (8i). White solid; FT IR (ATR, νmax, cm−1): 3059 (Ar–H), 2973, 2928 (C–H), 1629 (C
N), 1570 (CC), 783 (C–Cl); 1H NMR (400 MHz, DMSO, δ ppm): 3.32 (s, 2H, benzyl-CH2), 3.75 (s, 3H, imidazole-CH3), 7.47 (d, 2H, 2,4-dichlorophenyl-H, J = 8.4 Hz), 7.59 (s, 1H, 2,4-dichlorophenyl-H), 7.76 (d, 1H, benzimidazole-H, J = 8.4 Hz), 7.80–7.83 (m, 1H, Ar–H, benzimidazole-H), 7.90 (d, 1H, Ar–H, J = 8.8 Hz), 8.08 (t, 1H, Ar–H, J = 8.4 Hz), 8.16 (d, 1H, Ar–H, J = 8.4 Hz), 8.43 (s, 1H, Ar–H), 8.57 (s, 1H, benzimidazole-H); 13C NMR (125 MHz, DMSO, δ ppm): 30.82 (–CH3), 35.39 (–CH2), 111.59, 119.45, 121.94, 124.48, 126.82, 128.76, 129.80, 129.85, 130.99, 132.46, 133.28, 134.18, 136.45, 138.43, 142.40, 144.20, 153.68 (–CN), 166.77 (–CN), 174.77 (–CN); ESI-MS: 471.4 (M+ + H), 473.1 [(M + H) + 2]+, 475.4 [(M + H) + 4]+, 477.0 [(M + H) + 6]+; anal. calcd for C23H15Cl3N4O: C, 58.81; H, 3.22; N, 11.93. Found: C, 58.83; H, 3.23; N, 11.95.
4.1.6.10. 2-(2,4-Dichlorophenyl)-5-(3-(4-(methylsulfonyl)phenyl)-[1,2,4]-oxadiazol-5-yl)-1-methyl-1H-benzo[d]imidazole (8j). Light cream solid; FT IR (ATR, νmax, cm−1): 3081 (Ar–H), 2971, 2935 (C–H), 1382 (asym SO2), 1123 (sym SO2); 1H NMR (400 MHz, DMSO, δ ppm): 3.20 (s, 3H, CH3), 3.68 (s, 3H, CH3), 7.66–7.75 (m, 5H, Ar–H), 7.91 (d, 2H, Ar–H, J = 8.8 Hz), 8.09 (d, 2H, Ar–H, J = 8.4 Hz), 8.57 (s, 1H, Ar–H). 13C NMR (125 MHz, DMSO, δ ppm): 30.91 (CH3), 43.60 (CH2), 110.69, 121.45, 123.49, 124.14, 127.06, 127.86, 128.22, 129.42, 129.65, 133.72, 134.25, 136.00, 138.60, 139.25, 141.90, 143.18, 151.67 (–C
N), 157.31 (–CN), 163.95 (–CN); ESI-MS: 513.2 (M+ + H), 515.2 [(M + H) + 2]+, 417.1 [(M + H) + 4]+; anal. calcd for C23H16Cl2N4O3S: C, 55.32; H, 3.23; N, 11.22; S, 6.42. Found: C, 55.33; H, 3.24; N, 11.24; S, 6.43.
4.1.6.11. 2-(3-Chloro-2-fluorophenyl)-5-(3-(4-chlorophenyl)-[1,2,4]-oxadiazol-5-yl)-1-methyl-1H-benzo[d]imidazole (8k). White crystalline solid; FT IR (ATR, νmax, cm−1): 3034 (Ar–H), 2970, 2924 (C–H), 1618 (C
N), 1566 (CC), 1133 (C–F), 782 (C–Cl); 1H NMR (400 MHz, DMSO, δ ppm): 3.64 (s, 3H, –CH3), 7.35 (d, 2H, Ar–H, J = 7.6 Hz), 7.41–7.45 (m, 3H, 3-Cl-2-F phenyl ring), 7.55 (d, 1H, p-Cl phenyl ring, J = 8.4 Hz), 8.08 (d, 2H, p-Cl phenyl ring, J = 8.8 Hz), 8.18–8.20 (dd, 1H, J = 8.4 Hz and 1.2 Hz), 8.65 (s, 1H, benzimidazole-H); 13C NMR (125 MHz, DMSO, δ ppm): 30.47, 111.64, 118.74, 119.86, 122.61, 123.48, 124.40, 126.97, 128.57, 131.56, 132.26, 138.05, 142.09, 143.04, 155.89 (–CN), 156.73 (–CN), 162.86 (–CN); ESI-MS: 440.3 (M+ + H), 441.2 [(M + H) + 2]+, 443.2 [(M + H) + 4]+; anal. calcd for C22H13Cl2FN4O: C, 60.15; H, 2.98; N, 12.75. Found: C, 60.17; H, 2.97; N, 12.78.
4.1.6.12. 2-(3-Chloro-2-fluorophenyl)-5-(3-(pyridin-3-yl)-[1,2,4]-oxadiazol-5-yl)-1-methyl-1H-benzo[d]imidazole (8l). Light cream solid; FT IR (ATR, νmax, cm−1): 3039 (Ar–H), 2966, 2935 (C–H), 1608 (C
N), 1596 (CC), 1145 (C–F), 784 (C–Cl); 1H NMR (400 MHz, DMSO, δ ppm): 3.69 (s, 3H, –CH3), 7.42–7.46 (m, 3H, 3-Cl-2-F phenyl ring), 7.55 (d, 1H, Ar–H, J = 8.4 Hz), 7.62 (d, 1H, Ar–H, J = 8.4 Hz), 8.08 (s, 1H, Ar–H), 8.16–8.20 (m, 2H, Ar–H), 8.70 (s, 1H, benzimidazole-H); 13C NMR (125 MHz, DMSO, δ ppm): 30.87 (CH3), 112.63, 117.72, 118.86, 123.61, 124.48, 124.98, 126.97, 128.47, 129.53, 130.56, 132.66, 134.72, 138.55, 141.49, 143.04, 144.25, 155.89 (–CN), 156.73 (–CN), 162.86 (–CN); ESI-MS: 407.3 (M+ + H), 409.3 [(M + H) + 2]+; anal. calcd for C21H13ClFN5O: C, 62.15; H, 3.23; N, 17.26. Found: C, 62.17; H, 3.22; N, 17.29.
4.1.6.13. 2-(3-Chloro-2-fluorophenyl)-5-(3-(2-chlorophenyl)-[1,2,4]-oxadiazol-5-yl)-1-methyl-1H-benzo[d]imidazole (8m). Creamy white solid; FT IR (ATR, νmax, cm−1): 3059 (Ar–H), 2986, 2932 (C–H), 1628 (C
N), 1583 (CC), 1149 (C–F), 787 (C–Cl); 1H NMR (400 MHz, DMSO, δ ppm): 3.64 (s, 3H, –CH3), 7.35–7.39 (m, 3H, 3-Cl-2-F phenyl ring, J = 8 Hz), 7.50 (d, 1H, Ar–H, J = 8.4 Hz), 7.53–7.56 (m, 4H, Ar–H), 8.21–8.23 (dd, 1H, J = 8.8 Hz and 1.6 Hz), 8.65 (s, 1H, benzimidazole-H); 1H NMR (400 MHz, DMSO, δ ppm): 3.80 (s, 3H, –CH3), 7.19–7.87 (m, 9H, Ar–H), 8.71 (s, Ar–H); 13C NMR (125 MHz, DMSO, δ ppm): 31.27 (CH3), 110.84, 119.44, 119.56, 121.67, 123.46, 124.40, 125.93, 127.03, 129.57, 131.22, 132.16, 132.26, 132.88, 139.05, 142.09, 154.04, 155.89 (–CN), 156.03 (–CN), 163.81 (–CN); ESI-MS: 440.6 (M+ + H), 442.4 [(M + H) + 2]+, 444.7 [(M + H) + 4]+; anal. calcd for C22H13Cl2FN4O: C, 60.15; H, 2.98; N, 12.75. Found: C, 60.17; H, 2.97; N, 12.76.
4.1.6.14. 2-(3-Chloro-2-fluorophenyl)-5-(3-(3-chlorobenzyl)-[1,2,4]-oxadiazol-5-yl)-1-methyl-1H-benzo[d]imidazole (8n). White amorphous solid; FT IR (ATR, νmax, cm−1): 3059 (Ar–H), 2973, 2925 (C–H), 1602 (C
N), 1586 (CC), 1184 (C–F), 798 (C–Cl); 1H NMR (400 MHz, DMSO, δ ppm): 3.33 (s, 2H, benzyl-CH2), 3.72 (s, 3H, imidazole–CH3), 7.48–7.55 (m, 3H, 3-Cl-2-F phenyl ring-H), 7.60 (d, 1H, Ar–H, J = 8 Hz), 7.70–7.73 (m, 1H, Ar–H, benzimidazole-H), 7.78 (d, 1H, Ar–H, J = 8.4 Hz, benzimidazole-H), 7.90 (d, 1H, Ar–H, J = 8.8 Hz), 8.08 (t, 1H, Ar–H, J = 8.4 Hz), 8.43 (s, 1H, Ar–H), 8.61 (s, 1H, benzimidazole-H); 13C NMR (125 MHz, DMSO, δ ppm): 31.77 (CH3), 42.42 (CH2), 110.84, 118.64, 119.45, 120.52, 122.61, 124.46, 125.40, 126.93, 127.03, 130.57, 131.82, 132.16, 132.86, 132.98, 139.65, 142.59, 154.34, 155.89 (–CN), 157.03 (–CN), 162.81 (–CN); ESI-MS: 454.4 (M+ + H), 455.2 [(M + H) + 2]+, 457.2 [(M + H) + 4]+; anal. calcd for C23H15Cl2FN4O: C, 60.91; H, 3.34; N, 12.36. Found: C, 60.93; H, 3.36; N, 12.33.
4.1.6.15. 2-(3-Chloro-2-fluorophenyl)-5-(3-(4-(methylsulfonyl)phenyl)-[1,2,4]-oxadiazol-5-yl)-1-methyl-1H-benzo[d]imidazole (8o). FT IR (ATR, νmax, cm−1): 3029 (Ar–H), 2986, 2929 (C–H), 1618 (C
N), 1561 (CC), 1386 (asym SO2), 1188 (C–F), 1128 (sym SO2), 794 (C–Cl); 1H NMR (400 MHz, DMSO, δ ppm): 3.26 (s, 3H, CH3), 3.60 (s, 3H, CH3), 7.66–7.75 (m, 5H, Ar–H), 7.96 (d, 2H, Ar–H, J = 8.8 Hz), 8.12 (d, 2H, Ar–H, J = 8.4 Hz), 8.57 (s, 1H, Ar–H); 13C NMR (125 MHz, DMSO, δ ppm): 30.73 (CH3), 44.49 (CH3), 112.84, 118.68, 120.59, 122.51, 124.49, 125.70, 126.93, 128.03, 130.27, 131.87, 132.06, 132.86, 134.98, 139.62, 143.51, 154.34, 155.89 (–CN), 159.03 (–CN), 161.48 (–CN); ESI-MS: 483.2 (M+ + H), 484.2 [(M + H) + 2]+; anal. calcd for C23H16ClFN4O3S: C, 57.20; H, 3.34; N, 11.60; S, 6.64. Found: C, 57.21; H, 3.35; N, 11.61; S, 6.64.
4.1.6.16. 2-(2,3-Dichlorophenyl)-5-(3-(4-chlorophenyl)-[1,2,4]-oxadiazol-5-yl)-1-methyl-1H-benzo[d]imidazole (8p). Light yellow solid; FT IR (ATR, νmax, cm−1): 3077 (Ar–H), 2992, 2930 (C–H), 1623 (C
N), 1571 (CC), 778 (C–Cl); 1H NMR (400 MHz, DMSO, δ ppm): 3.68 (s, 3H, –CH3), 7.42 (d, 1H, benzimidazole-H, J = 8 Hz), 7.45 (d, 2H, 4-chlorophenyl, J = 8.4 Hz), 7.50 (d, 2H, 4-chlorophenyl, J = 8.4 Hz), 7.68 (d, 1H, 2,3-dichlorophenyl, J = 8.4 Hz), 7.83 (t, 1H, 2,3-dichlorophenyl, J = 8.4 Hz), 8.09 (d, 1H, 2,3-dichlorophenyl, J = 8.8 Hz), 8.15–8.18 (dd, 1H, benzimidazole-H, J = 8.4 Hz and 1.6 Hz), 8.66 (s, 1H, benzimidazole-H); 13C NMR (125 MHz, DMSO, δ ppm): 31.30 (–CH3), 112.55, 119.75, 121.77, 123.36, 125.61, 127.84, 128.86, 129.15, 129.88, 133.18, 137.22, 154.60 (–CN), 169.16 (–CN), 176.81 (–CN); ESI-MS: 455.5 (M+ + H), 457.1 [(M + H) + 2]+, 459.1 [(M + H) + 4]+, 461.0 [(M + H) + 6]+; anal. calcd for C23H15Cl2FN4O: C, 57.98; H, 2.88; N, 12.29. Found: C, 57.97; H, 2.89; N, 12.31.
4.1.6.17. 2-(2,3-Dichlorophenyl)-5-(3-(pyridin-3-yl)-[1,2,4]-oxadiazol-5-yl)-1-methyl-1H-benzo[d]imidazole (8q). White solid; FT IR (ATR, νmax, cm−1): 3055 (Ar–H), 2993, 2927 (C–H), 1629 (C
N), 1579 (CC), 773 (C–Cl); 1H NMR (400 MHz, DMSO, δ ppm): 3.69 (s, 3H, –CH3), 7.40 (d, 1H, 2,3-dichlorophenyl, J = 8.4 Hz), 7.50 (t, 1H, 2,3-dichlorophenyl, J = 8.4 Hz), 7.55 (d, 1H, 2,3-dichlorophenyl, J = 8.4 Hz), 7.59 (d, 1H, Ar–H, J = 8 Hz), 7.63 (d, 1H, Ar–H, J = 7.6 Hz), 8.08–8.12 (m, 3H, Ar–H), 8.17–8.20 (dd, 1H, benzimidazole-H, J = 8.4 Hz and 1.2 Hz), 8.64 (s, 1H, benzimidazole-H); 13C NMR (125 MHz, DMSO, δ ppm): 30.20 (–CH3), 109.99, 116.57, 118.24, 121.98, 122.36, 124.62, 125.41, 127.74, 128.36, 129.45, 129.88, 130.31, 133.18, 137.22, 138.20, 154.64 (–CN), 169.16 (–CN), 175.59 (–CN); ESI-MS: 455.1 (M+ + H), 457.5 [(M + H) + 2]+; anal. calcd for C21H13Cl2N5O: C, 59.73; H, 3.10; N, 16.79. Found: C, 59.72; H, 3.09; N, 16.80.
4.1.6.18. 2-(2,3-Dichlorophenyl)-5-(3-(2-chlorophenyl)-[1,2,4]-oxadiazol-5-yl)-1-methyl-1H-benzo[d]imidazole (8r). White solid; FT IR (ATR, νmax, cm−1): 3057 (Ar–H), 2993, 2930 (C–H), 1621 (C
N), 1572 (CC), 779 (C–Cl); 1H NMR (400 MHz, DMSO, δ ppm): 3.64 (s, 3H, –CH3), 7.34 (d, 1H, 2,3-dichlorophenyl, J = 8.4 Hz), 7.52–7.55 (m, 4H, Ar–H), 7.77 (d, 1H, benzimidazole-H, J = 8 Hz), 7.87 (t, 1H, 2,3-dichlorophenyl, J = 8.4 Hz), 8.04 (d, 1H, 2,3-dichloro phenyl-H, J = 8.6 Hz), 8.22–8.25 (dd, 1H, J = 8.4 Hz and 1.2 Hz), 8.65 (s, 1H, benzimidazole-H); 13C NMR (125 MHz, DMSO, δ ppm): 30.56 (–CH3), 111.59, 117.45, 120.94, 124.36, 125.82, 126.76, 128.80, 129.85, 129.99, 133.18, 135.32, 136.22, 137.46, 140.39, 142.36, 153.70 (–CN), 166.76 (–CN), 134.71 (–CN); ESI-MS: 455.1 (M+ + H), 457.1 [(M + H) + 2]+, 459.5 [(M + H) + 4]+, 461.0 [(M + H) + 6]+; anal. calcd for C22H13Cl3N4O: C, 57.98; H, 2.88; N, 12.29. Found: C, 57.99; H, 2.89; N, 12.28.
4.1.6.19. 2-(2,3-Dichlorophenyl)-5-(3-(3-chlorobenzyl)-[1,2,4]-oxadiazol-5-yl)-1-methyl-1H-benzo[d]imidazole (8s). White solid; FT IR (ATR, νmax, cm−1): 3059 (Ar–H), 2973, 2928 (C–H), 1629 (C
N), 1575 (CC), 814 (C–Cl); 1H NMR (400 MHz, DMSO, δ ppm): 3.32 (s, 2H, benzyl-CH2), 3.75 (s, 3H, –CH3), 7.47 (d, 1H, dichlorophenyl, J = 8 Hz), 7.59 (t, 1H, 2,3-dichlorophenyl, J = 8 Hz), 7.76 (d, 1H, benzimidazole-H, J = 8.4 Hz), 7.80 (d, 1H, dichlorophenyl, J = 8 Hz), 7.85–7.88 (m, 1H, Ar–H, benzimidazole-H), 7.96 (d, 1H, Ar–H, J = 8.8 Hz), 8.08 (t, 1H, Ar–H, J = 8.4 Hz), 8.16 (d, 1H, Ar–H, J = 8.4 Hz), 8.43 (s, 1H, Ar–H), 8.57 (s, 1H, benzimidazole-H); 13C NMR (125 MHz, DMSO, δ ppm): 32.82 (–CH3), 39.39 (–CH2), 111.59, 119.45, 121.44, 124.48, 126.72, 129.76, 129.80, 129.85, 131.99, 132.46, 133.28, 134.18, 137.45, 138.43, 142.46, 144.54, 153.69 (–CN), 166.77 (–CN), 175.77 (–CN); ESI-MS: 471.4, 473.1 [(M + H) + 2]+, 475.4 [(M + H) + 4]+, 477.0 [(M + H) + 6]+; anal. calcd for C23H15Cl3N4O: C, 58.81; H, 3.22; N, 11.93. Found: C, 58.83; H, 3.24; N, 11.92.
4.1.6.20. 2-(2,3-Dichlorophenyl)-5-(3-(4-(methylsulfonyl)phenyl)-[1,2,4]-oxadiazol-5-yl)-1-methyl-1H-benzo[d]imidazole (8t). Light orange solid; FT IR (ATR, νmax, cm−1): 3071 (Ar–H), 2971, 2935 (C–H), 1386 (asym SO2), 1113 (sym SO2), 803 (C–Cl); 1H NMR (400 MHz, DMSO, δ ppm): 3.24 (s, 3H, –CH3), 3.69 (s, 3H, –CH3), 7.68–7.77 (m, 5H, Ar–H), 7.98 (d, 2H, Ar–H, J = 8.8 Hz), 8.18 (d, 2H, Ar–H, J = 8.4 Hz), 8.60 (s, 1H, Ar–H). 13C NMR (125 MHz, DMSO, δ ppm): 30.95 (CH3), 43.63 (CH2), 111.69, 122.45, 123.49, 124.14, 127.06, 127.86, 128.22, 129.45, 129.64, 133.71, 134.25, 136.04, 138.67, 139.25, 141.90, 144.18, 152.67 (–C
N), 158.31 (–CN), 164.95 (–CN); ESI-MS: 513.1 (M+ + H), 515.5 [(M + H) + 2]+, 417.4 [(M + H) + 4]+; anal. calcd for C23H16Cl2N4O3S: C, 55.32; H, 3.23; N, 11.22; S, 6.42. Found: C, 55.34; H, 3.25; N, 11.24; S, 6.44.
4.2. Antimicrobial activity
The MIC of the samples in DMSO was determined by using different concentrations of compounds in brain heart infusion broth for bacteria and fungi by the macro dilution method. The lowest concentration of the sample in DMSO inhibiting the visible growth of microorganisms was considered as MIC. Here the broth dilution test was used to determine MIC of samples 8a–t. Freshly prepared nutrient broth was used as diluent. The 24 h old cultures of the test bacteria S. aureus, E. faecalis, E. coli and K. pneumoniae and the test fungi C. albicans and A. fumigatus were diluted 100-fold in nutrient broth (NB) (100 μL bacterial cultures in 10 mL NB). A stock solution of the synthesized compounds with concentration of 1 mg mL−1 was prepared in DMSO. Different concentrations of the test samples (0.2, 0.4, 0.8, 1.6, 3.125, 6.25, 12.5, 25, 50 and 100 μg mL−1) were added to the test tubes containing the bacterial and fungal cultures. All the tubes were incubated at 37 °C for 24 h for bacteria and at 28 °C for 48 h for fungi. The tubes were examined for visible turbidity and using NB as a control. Control without test samples and with solvent (DMSO) was assayed simultaneously. The lowest concentration that inhibited visible growth of the tested organisms was recorded as MIC.4.3. In vitro anti-tubercular activity
Anti-tubercular activity was evaluated against Mycobacterium tuberculosis H37Rv by Microplate Alamar Blue Assay (MABA). In the presence of cellular metabolism, the colour of Alamar Blue changes from a fully oxidized, non-fluorescent blue to a fully reduced, fluorescent pink colour. This methodology is nontoxic, uses a thermally stable reagent and shows good correlation with the BACTEC radiometric method. Middlebrook 7H9 broth base with Middlebrook OADC growth supplement was used as medium. 200 μL of sterile deionized water was added to all outer perimeter wells of sterile 96-well plate to minimize evaporation of medium in the test wells during incubation. The 96-well plate received 100 μL of the Middlebrook 7H9 broth. 10 mg of each compound was dissolved in 10.0 mL of DMSO separately and was further diluted with DMSO to make 100 μg mL−1 and 10 μg mL−1 stock solution. Using the above stock solution, serial dilutions of 0.2 μg mL−1, 0.8 μg mL−1, 1.6 μg mL−1, 3.12 μg mL−1, 6.25 μg mL−1, 12.5 μg mL−1, 25 μg mL−1, 50 μg mL−1 and 100 μg mL−1 were made with DMSO directly on the plate. Further the positive control was prepared by inoculating the test organism with medium containing standard drugs and a negative control was prepared by inoculating test organism with medium containing only DMSO. 50 μL of DMSO was used as control. Medium without test compound/DMSO was also inoculated with the test organism to check whether the media will support the growth of the tubercle bacilli or not, and no mycobacteria growth was observed. The abovementioned wells were inoculated with M. tuberculosis H37Rv strain. Plates were covered and sealed with Parafilm and incubated at 37 °C for five days. After this time, 25 μL of freshly prepared 1:1 mixture of Alamar Blue reagent and 10% Tween 80 was added to the plate and incubated for 24 h. A blue color in the well was interpreted as no bacterial growth, and pink color was scored as growth.
4.4. Hemolytic assay
Human blood was collected in an EDTA (2 mg mL−1)-containing vacutainer. Plasma and buffy coat were removed from the EDTA-blood by centrifugation at 800 × g for 10 min. Later, the erythrocytes were washed three times by using normal saline (0.9%) and resuspended in saline to 5% erythrocytes suspension. The cells were incubated for 1 h at 37 °C in the presence of test compounds. After the incubation, the samples were centrifuged at 800 × g for 10 min and the absorbance was read using a UV spectrophotometer. 2% Triton X-100 (Sigma-Aldrich, St Louis, USA) served as positive control. Phosphate buffered saline served as negative control. The absorbance of haemoglobin release was measured at 540 nm and is expressed as % of Triton X-100 induced hemolysis. The result was calculated by using the following formula:| % Hemolysis = [(absorbance of sample) − (absorbance of blank)/highest absorbance of positive control] × 100 |
Each concentration had triplicate values. HC50 was determined for each triplicate absorbance measurement and average of HC50 was plotted against concentration fitted with a sigmoid plot. From the curve, concentration corresponding to 50% hemolysis was determined.
4.5. In vitro cytotoxic activity
Human embryonic kidney cell line HEK 293 was procured from NCCS, Pune. The cytotoxicity effects of 8k, 8n, 8p and 8r were studied by MTT assay. The cells were cultured using Eagle's minimum essential medium (MEM) supplemented with 10% fetal bovine serum, penicillin (100 U mL−1) and streptomycin (100 μg mL−1) and grown in 25 cm2 tissue culture flasks at 37 °C with 5% CO2 incubator (Galaxy 170S, New Brunswick). After attaining 70–80% confluence, cells were trypsinized and seeded to 96-well plates in 100 μL of MEM (5000 cells/well). After 24 h, different concentrations of drugs were dissolved in DMSO and added to 96-well plates and incubated for 24 h at standard condition. Untreated cells represented a control group. Four replicates were set up for each test group. After completion of the incubation time, 20 μL of MTT dye (5 mg mL−1 in PBS) was added to all wells; the plate was sealed with aluminium foil and incubated for 4 h in an incubator. After 4 h, 100 μL of DMSO was added to all the wells and mixed by uniform shaking. Using a multi-plate reader (LisaScan®EM) the absorbance was recorded at 570 nm. The percentage of viable cells was calculated using the following formula:| Percentage of viability cells = (test sample-blank/control-blank) × 100 |
Each concentration had triplicate values and the average value was plotted against concentration by fitting with a sigmoid plot. From the curve, the concentration corresponding to 50% cell viability was noted.
Acknowledgements
The authors are grateful to UGC-SAP and DST-PURSE for providing financial assistance. We are grateful to USIC Mangalore University and SAIF Cochin for providing spectral analyses. We also gratefully acknowledge The Director and Mr Vishwanatha U, SDM Centre for Research in Ayurveda, Udupi for their help in carrying out the cytotoxicity study.References
- R. G. Ducati, A. Ruffino-Netto, L. A. Basso and D. S. Santos, Mem. Inst. Oswaldo Cruz, 2006, 101, 697–714 CrossRef CAS PubMed.
- A. Kapur and A. D. Harries, Diabetes Res. Clin. Pract., 2013, 101, 10–19 CrossRef PubMed.
- T. Fujita, M. Endo, Y. Gu, T. Sato and N. Ohmagari, J. Infect. Chemother., 2014, 20, 213–216 CrossRef PubMed.
- A. Doblas, F. Alcaide, N. Benito, M. Gurgui and T. Cisneros, Enfermedades Infecciosas y Microbiología Clínica, 2012, 30, 34–39 CrossRef PubMed.
- L. G. Dover and G. D. Coxon, J. Med. Chem., 2011, 54, 6157–6165 CrossRef CAS PubMed.
- A. Kumar, S. Srivastava, G. Gupta, V. Chaturvedi, S. Sinha and R. Srivastava, ACS Comb. Sci., 2011, 13, 65–71 CrossRef CAS PubMed.
- K. E. A. Lougheed, D. L. Taylor, S. A. Osborne, J. S. Bryans and R. S. Buxton, Tuberculosis, 2009, 89, 364–370 CrossRef CAS PubMed.
- G. N. Tew, R. W. Scott, M. L. Klein and W. F. Degrado, Acc. Chem. Res., 2010, 43, 30–39 CrossRef CAS PubMed.
- M. M. Konai and J. Halder, ACS Infect. Dis., 2015, 1, 469–478 CrossRef CAS.
- K. V. Shashidhara, K. B. Rao, P. Kushwaha, R. K. Modukuri, P. Singh, I. Soni, P. K. Shukla, S. Chopra and M. Pasupuleti, ACS Med. Chem. Lett., 2015, 6, 809–813 CrossRef PubMed.
- W. C. C. Yang, V. W. L. Ng, Y. H. J. Cheng, Y. W. Tong, D. J. Coady, W. Fan, J. L. Hedrick and Y. Y. Yang, Macromolecules, 2013, 46, 8797–8807 CrossRef.
- M. M. Konai, C. Ghosh, V. Yarlagadda, S. Samaddar and J. Haldar, J. Med. Chem., 2014, 57, 9409–9423 CrossRef CAS PubMed.
- H. A. Barker, R. D. Smyth, H. Weissbach, J. I. Toohey, J. N. Ladd and B. E. Volcani, J. Biol. Chem., 1960, 235, 480–488 CAS.
- R. C. Wende and P. R. Schreiner, Green Chem., 2012, 14, 1821–1849 RSC.
- M. Rashid, A. Husain and R. Mishra, Eur. J. Med. Chem., 2012, 54, 855–866 CrossRef CAS PubMed.
- A. Hamze, J. F. Hernandez, P. Fulcrand and J. Martinez, J. Org. Chem., 2003, 68, 7316–7321 CrossRef CAS PubMed.
- N. R. Thimmegoda, S. S. Nanjunda, K. C. S. Ananda, G. W. Yip and K. S. Rangppa, Bioorg. Med. Chem. Lett., 2008, 18, 432–435 CrossRef PubMed.
- A. S. Kiselyov, M. N. Semenova, N. B. Chernyshova, A. Leitao, A. V. Samet, K. A. Kislyi, M. M. Raihstat, T. Oprea, H. Lemcke, M. Lantow, D. G. Weiss, N. N. Ikizalp, S. A. Kuznetsov and V. V. Semenov, Eur. J. Med. Chem., 2010, 45, 1683–1697 CrossRef CAS PubMed.
- S. M. Wignall, N. S. Gray, Y. T. Chang, L. Juarez, R. Jacob, A. Burlingame, P. G. Schultz and R. Heald, Chem. Biol., 2004, 11, 135–146 CrossRef CAS PubMed.
- Z. Liu, Z. Zhou, W. Tian, X. Fan, D. Xue, L. Yu, Q. Yu and Y.-Q. Long, ChemMedChem, 2012, 7, 680–693 CrossRef CAS PubMed.
- F. Mollinedo and C. Gajate, Apoptosis, 2003, 8, 413–450 CrossRef CAS PubMed.
- J. Pérez-Villanueva, A. Hernández-Campos, L. Yépez-Mulia, C. Méndez-Cuesta, O. Méndez-Lucio, F. Hernández-Luis and R. Castillo, Bioorg. Med. Chem. Lett., 2013, 23, 4221–4224 CrossRef PubMed.
- H. Zhu, J. Zhang, N. Xue, Y. Hu, B. Yang and Q. He, Invest. New Drugs, 2010, 28, 493 CrossRef CAS PubMed.
- R. V. Shingalapur, K. M. Hosamani and R. S. Keri, Eur. J. Med. Chem., 2009, 44, 4244–4248 CrossRef CAS PubMed.
- V. Klimesova, J. Koci, K. Waisser and J. Kaustova, Il Farmaco, 2002, 57, 259–265 CrossRef CAS PubMed.
- M. Pieroni, S. K. Tipparaju, S. Lun, Y. Song, A. W. Sturm, W. R. Bishai and A. P. Kozikowski, ChemMedChem, 2011, 6, 334–342 CrossRef CAS PubMed.
- V. A. Kumar, K. J. Seetharamappa, U. Katrahalli and K. G. Bhat, Eur. J. Med. Chem., 2014, 79, 194–202 CrossRef PubMed.
- M. Goebel, G. Wolber, P. Markt, B. Staels, T. Unger, U. Kintscher and R. Gust, Bioorg. Med. Chem., 2010, 18, 5885–5895 CrossRef CAS PubMed.
- H. Dogan, A. Daran, S. Rollas, G. Sener, M. K. Uysal and D. Gulen, Bioorg. Med. Chem., 2002, 10, 2893–2898 CrossRef CAS PubMed.
- R. M. Srivastava, A. Lima, O. S. Viana, M. J. C. Silva, M. T. J. A. Catanho and J. O. F. Morais, Bioorg. Med. Chem., 2003, 11, 1821–1827 CrossRef CAS PubMed.
- P. Afiatpour, R. M. Srivastava, M. L. Oliveira and E. J. Barreiro, Braz. J. Med. Biol. Res., 1994, 27, 1403–1406 CAS.
- N. M. M. Bezerra, S. P. de Oliveira, R. M. Srivastava and J. R. de Silva, Il Farmaco, 2005, 60, 955–960 CrossRef CAS PubMed.
- M. T. H. Khan, M. I. Choudhary, K. M. Khan, M. Rani and A. U. Ahman, Bioorg. Med. Chem., 2005, 13, 3385–3395 CrossRef CAS PubMed.
- S. Manfredini, I. Lampronti, S. Vertuani, N. Solaroli, M. Recanatini, D. Bryan and M. McKinney, Bioorg. Med. Chem., 2000, 8, 1559–1566 CrossRef CAS PubMed.
- J. W. Clitherow, P. Beswick, W. J. Irving, D. I. C. Scopes, J. C. Barnes, J. Clapham, J. D. Brown, D. J. Evans and A. G. Hayes, Bioorg. Med. Chem. Lett., 1996, 6, 833–838 CrossRef CAS.
- J. B. Acri, G. Wong and J. M. Witkin, Eur. J. Pharmacol., 1995, 278, 213–223 CrossRef CAS PubMed.
- D. Kim, L. M. Wang, M. Beconi, G. J. Eiermann, M. H. Fisher, H. He, G. J. Hickey, J. E. Kowalchick, B. Leiting, K. Lyons, F. Marsilio, M. E. McCann, R. A. Patel, A. Petrov, G. Scapin, S. B. Patel, R. S. Roy, J. K. Wu, M. J. Wyvratt, B. B. Zhang, L. Zhu, N. A. Thornberry and A. E. Weber, J. Med. Chem., 2005, 48, 141–151 CrossRef CAS PubMed.
- K. Vasantha, B. Poojary, A. Prathibha and N. Shruthi, Synth. Commun., 2014, 44, 3414–3425 CrossRef.
- K. Vasantha, G. B. Swamy, M. V. Rai, B. Poojary, V. R. Pai, N. Shruthi and B. Mahima, Bioorg. Med. Chem. Lett., 2015, 25, 1420–1426 CrossRef CAS PubMed.
- G. Aguirre, M. Bolani, H. Cerecetto, A. Gerpe, M. Gonzalez, Y. F. Sainz, A. Denicola, C. O. de Ocariz, J. J. Nogal, D. J. Montero and A. Escario, Arch. Pharm., 2004, 337, 259–270 CrossRef CAS PubMed.
- National Committee for Clinical Laboratory Standards, Methods for Dilution Antimicrobial Susceptibility for Bacteria Grown Aerobically, Approved Standard, National Committee for Clinical Laboratory Standards, Villanova, A, 1985 Search PubMed.
- L. A. Collins and S. G. Franzblau, Antimicrob. Agents Chemother., 1997, 41, 1004–1009 CAS.
- A. Leo, C. Hansch and D. Elkins, Chem. Rev., 1971, 71, 525–616 CrossRef CAS.
- P. K. Ranjith, P. Rajeesh, K. R. Haridas, N. K. Susanta, T. N. Guru Row, R. Rishikesan and N. S. Kumari, Bioorg. Med. Chem. Lett., 2013, 23, 5228–5234 CrossRef CAS PubMed.
- M. M. Konai, C. Ghosh, V. Yarlagadda, S. Samaddar and J. Haldar, J. Med. Chem., 2014, 57, 9409–9423 CrossRef CAS PubMed.
- L. L. Gundersen, J. Nissen-Meyer and B. Spilsberg, J. Med. Chem., 2002, 45, 1383–1386 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3–26 CrossRef CAS PubMed.
- M. A. A. El-Sayed, N. I. Abdel-Aziz, A. A. M. Abdel-Aziz, A. S. El-Azab and K. E. H. El-Tahir, Bioorg. Med. Chem., 2012, 20, 3306–3316 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary data contain scanned spectra of compounds and dose response curve of mitomycin C against normal cell line. See DOI: 10.1039/c5ra23282a |
| This journal is © The Royal Society of Chemistry 2016 |