DOI:
10.1039/C5RA25684D
(Paper)
RSC Adv., 2016,
6, 8317-8328
Clickable trimethylguanosine cap analogs modified within the triphosphate bridge: synthesis, conjugation to RNA and susceptibility to degradation†
Received
2nd December 2015
, Accepted 10th January 2016
First published on 14th January 2016
Abstract
The trimethylguanosine (m3G) cap present at the 5′ end of small nuclear RNAs (snRNAs) has been proposed as an effective nuclear localization signal (NLS) for nucleus-targeting therapeutics such as antisense oligonucleotides. To provide novel tools for studies on m3G-mediated transport and m3G degradation, we synthesized a series of novel m3G cap analogs that combine modifications potentially affecting its activity as an NLS and stability in vivo with a modification enabling simple conjugation to biomolecules. The synthesized dinucleotide m3G analogs carry a single phosphate-modification (phosphorothioate, methylenebisphosphonate or imidodiphosphate) at the selected position of the triphosphate bridge in order to increase their resistance to enzymatic cleavage and a (2-azidoethyl)-carbamoylmethyl group at the 2′-position of adenosine as a second nucleotide to enable conjugation to alkyne-containing biomolecules by copper catalyzed azide–alkyne cycloaddition (CuAAC). The susceptibility of m3G cap analogs to non-specific and specific degradation was studied in fetal bovine serum and in an in vitro decapping assay with hNUDT16 enzyme, respectively. The susceptibility of m3G cap analogs to hNUDT16 mediated decapping was also determined after their CuAAC-mediated conjugation to a model oligonucleotide bearing a 5′-alkyne group. Depending on the type and the position of introduced modifications, they modulate the susceptibility to specific and non-specific degradation of conjugated molecules to various extent, with O to NH substitution at the α/β position providing the greatest m3G stability against hNUDT16.
Introduction
Hypermethylated 2,2,7-trimethylguanosine (m3G) cap structures are present at the 5′ end of small nuclear RNAs that program mRNA splicing (U1, U2, U4 and U5 snRNAs) and some nucleolar RNAs (snoRNAs).1,2 For snRNAs the m3G cap is formed post-transcriptionally by the cytoplasmic enzyme Tgs1, which catalyzes two successive methyl additions at the N2 position of the m7G cap in premature transcripts.3,4 Then, m3G-capped snRNAs are recognized by snurportin, an adaptor protein that binds to importin β. This interaction mediates the import of matured snRNA back to the nucleus.5–8 It has been shown that the presence of an m3G cap is required for efficient nuclear importing of snRNAs and for proper mRNA splicing.8 Therefore, many m3G cap analogs have been synthesized and applied in numerous biophysical and biological studies on the transport and function of small RNAs.9–11 Importantly, they have been also proposed as a nuclear localization signal (NLS) for delivery of the splice-correcting antisense oligonucleotides (ONs)12 designed to act inside the nucleus. As shown there, m3G-capped oligonucleotides were efficiently imported into the nucleus, even as a part of large bioconjugates, and promoted splicing in model systems more efficiently than uncapped ones,12 thus indicating their potential of use in experimental therapies for genetic disorders such as Duchenne Muscular Dystrophy (DMD)13 or Spinal Muscular Atrophy (SMA).14,15 However, for future developments it would be beneficial to design m3G cap analogs that not only interact with snurportin but can also be easily incorporated into RNA and are resistant to specific and non-specific enzymatic degradation. The unmodified m3G cap structure is potentially susceptible to degradation by extra- and intracellular pyrophosphatases that cleave the triphosphate bridge between α/β or β/γ phosphates. Little is known about degradation pathways specific for m3G-capped RNAs, but recent studies reveal involvement of NUDIX pyrophosphatases in the decapping process.16 It has been shown that m3G-capped RNAs are decapped by the NUDT16 enzyme in vitro. Recently, also Dcp2, known primarily as a major decapping enzyme targeting monomethylguanosine (m7G) capped mRNAs, have been shown to hydrolyze m3G-capped snRNAs as a part of a quality control mechanism.16,17 Both enzymes cleave the triphosphate bridge of the cap between α and β phosphates to release methylated GDP and 5′-phosphorylated RNA.16,17 Several modifications have been recently identified that protect m3G and m7G caps against degradation by hNUDT16 and Dcp2, respectively, as well as by other cap-specific and non-specific enzymes.18–25 For example, introduction of β-phosphorothioate,20 α,β-methylenebisphosphonate26 or α,β-imidodiphosphate22 groups into the triphosphate bridge of the m7G cap significantly improves the cellular half-life of exogenously delivered capped-mRNAs.19,27,28 In this work, we aimed at combining the phosphate modifications of m3G cap with another beneficial feature, i.e. possibility of facile conjugation to oligonucleotides using click chemistry29 as accomplished with unmodified caps.30 Therefore, we synthesized a set of six novel phosphate-modified m3G cap analogues (Fig. 1) that are equipped with 2-(azidoethyl)carbamoylmethyl handle at the 2′-position of the ribose moiety, which enables efficient conjugation of m3G caps to alkyne modified RNAs by copper catalyzed azide–alkyne cycloaddition (CuAAC)30 (Fig. 1).
 |
| | Fig. 1 Structures of m3G-cap analogs synthesized in this study. * a and b refer to either of the two P-diastereomers of a given compound, D1 and D2. D1 denotes the isomer eluting faster from a reversed-phased HPLC column. | |
The analogs were modified at various positions of the 5′,5′-triphosphate bridge by replacing either one of the bridging oxygens with CH2 or NH groups, or one of the non-bridging oxygens with sulfur. All synthesized dinucleotide m3G cap analogs were then used to study unspecific degradation in fetal bovine serum (FBS). To determine the influence of the modifications on susceptibility to decapping enzymes, the m3G caps were then attached to model short RNA using “click chemistry” approach and studied for their susceptibility to decapping by recombinant hNUDT16 in vitro.
Results and discussion
Synthesis of modified m3G cap analogs 1–7
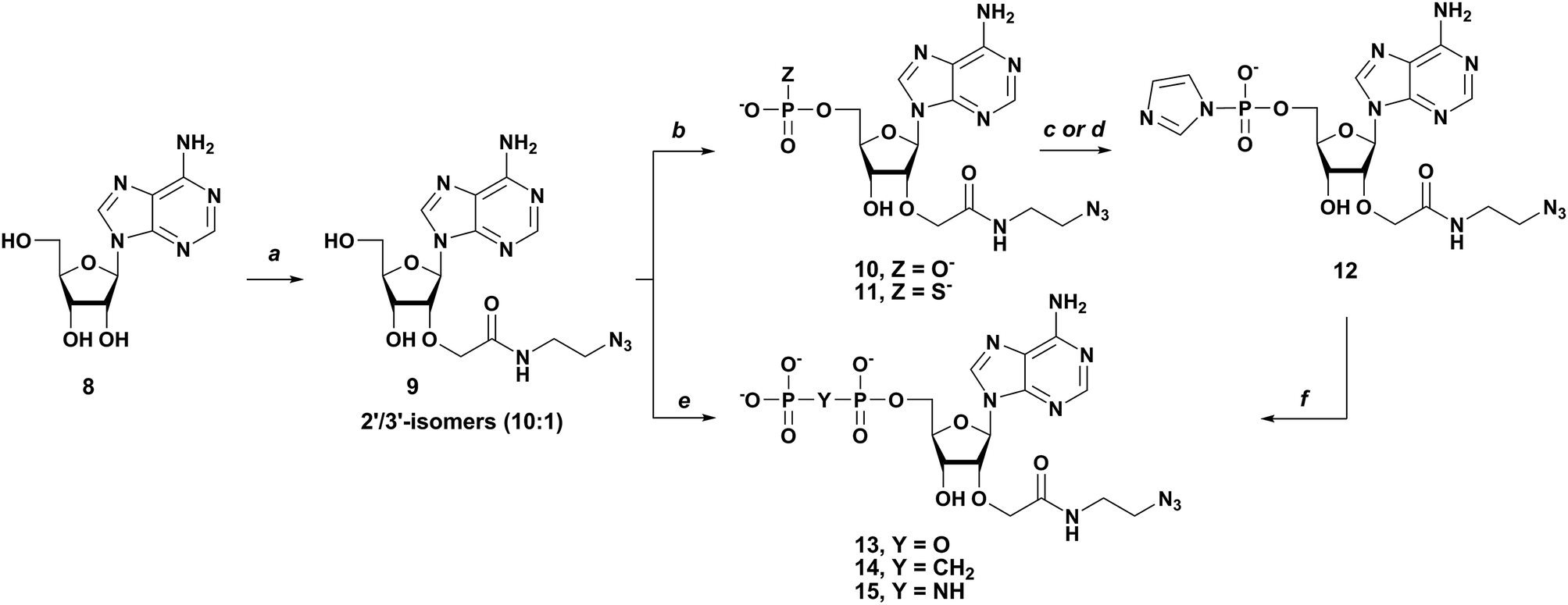
The syntheses of six phosphate-modified m3G cap analogs functionalized with (2-azidoethyl)carbamoylmethyl group at the 2′-position of adenosine together with an unmodified parent compound described previously are shown in Schemes 1–3.30,31 The synthesis was accomplished by coupling two mononucleotide units taking advantage of P-imidazolide chemistry. The key starting materials in the synthesis of all m3G cap analogs were an adenosine-derived building block bearing a “clickable” azido linker at 2′-position and a m3G-derived building block, one of which had to be modified in the phosphate moiety and the other activated with imidazole.30 2′-O-(N-(2-Azidoethyl)carbamoyl)methyl adenosine 9 was synthesized by a minor modification of the previously described procedure30 to avoid 5′-OH protection, since we observed that MMTr group removal in acidic conditions is accompanied with partial degradation of the amide bond yielding also 2′-O-(carboxymethyl)adenosine. Alkylation of unprotected adenosine (8) (Scheme 1) was performed in a one-pot two-step procedure involving NaH and allyl bromoacetate treatment followed by addition of 2-azidoethylamine in DMF to provide the desired 2′-alkylated product 930 with a good regioselectivity (2′![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3′ as 10:1) and in 80–90% yield. Next, modified Yoshikawa's phosphorylation procedure32 was employed to convert 9 into its 5′-monophosphate (10)30 or 5′ monophosphorothioate (11) using POCl3 or PSCl3, to afford compounds 10 and 11 in, respectively, 85% and 65% isolated yields after ion-exchange chromatography (IEC) (Scheme 1).
:3′ as 10:1) and in 80–90% yield. Next, modified Yoshikawa's phosphorylation procedure32 was employed to convert 9 into its 5′-monophosphate (10)30 or 5′ monophosphorothioate (11) using POCl3 or PSCl3, to afford compounds 10 and 11 in, respectively, 85% and 65% isolated yields after ion-exchange chromatography (IEC) (Scheme 1).
 |
| | Scheme 1 Synthesis of 2′-O-(N-(2-azidoethyl)carbamoyl)methyladenosine (9) and its 5′-phosphate derivatives (10–15) (a) (i) NaH (4 equiv.), dry DMF, 10 min, r.t; allyl bromoacetate (2 equiv.), r.t, 1 h; (ii) 2-Azidoethylamine (3 equiv.), 24 h, r.t.; (b) (i) PXCl3 (X = O, S) (3 equiv.), PO(OMe)3, 3–4 h, 0 °C, (ii) NaHCO3aq (9 equiv.); (c) (i) imidazole (10 equiv.), 2,2′-dithiopyridine (3 equiv.), TEA (3 equiv.), PPh3 (3 equiv.); DMF, 24 h, r.t.; (ii) NaClO4, acetone; (d) in situ, CDI (5 equiv.), DMF, MW (40 °C, 5 W, 20 min), (e) (i) Cl2POCH2POCl2 (3 equiv.) for 14 or Cl3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NP(O)Cl2, (3 equiv.) PO(OMe)3 for 15, 0 °C, 6–8 h; (ii) NaHCO3aq.(15 equiv.), (e) PO43−/TEA+ (2 equiv.) for 13, ZnCl2 (8 equiv.), DMF, 6 h, r.t. NP(O)Cl2, (3 equiv.) PO(OMe)3 for 15, 0 °C, 6–8 h; (ii) NaHCO3aq.(15 equiv.), (e) PO43−/TEA+ (2 equiv.) for 13, ZnCl2 (8 equiv.), DMF, 6 h, r.t. | |
 |
| | Scheme 2 Synthesis of dinucleotide m3G cap analogs 1–3 and 7; (a) ZnCl2, (10 equiv.), DMF; (b) ZnCl2, (10 equiv.) DMF. | |
 |
| | Scheme 3 Synthesis of dinucleotide m3G-cap analogs 4–6; (a) imidazole (10 equiv.), 2,2′-dithiodipyridine (3 equiv.), TEA (3 equiv.), PPh3 (3 equiv.); DMF, 24 h, r.t.; (ii) NaClO4, acetone or (a) CDI (5 equiv.), DMF, MW (40 °C, 5 W, 20 min), (b) H2O (8 equiv.) 5 min, (ii) ZnCl2 (10 equiv.). | |
Similarly, 2′-O-(N-(2-azidoethyl)carbamoyl)methyladenosine 5′-methylene-(bisphosphonate) (14) or imidodiphosphate (15) were obtained by reacting 9 with methylenebis(phosphonic chloride) or dichlorophosphorylphosphorimidoyltrichloride.25,33 Compounds 14 and 15 were obtained after IEC purification in 60% and 50% yields, respectively. The synthesis of the reactive nucleotide P-imidazolide 12 was achieved either by in situ activation with 1,1′-carbonyldiimidazole (CDI) accelerated by microwave irradiation in DMF34,35 or by the Mukaiyama–Hashimoto method employing imidazole, 2,2′ dithiodipyridine, triethylamine and triphenylphosphine in DMF and subsequent precipitation by sodium perchlorate solution in acetone.36 Noteworthy, we did not observe any azide reduction using PPh3. The synthetic route to m3G cap analogs unmodified 130 or bearing a modification at the α/β position of triphosphate bridge 2–3, and 7a and b is depicted in Scheme 2. Compounds bearing the O to CH2 or NH (2 and 3) substitutions at the α/β position of the 5′,5′-triphosphate bridge (Scheme 2) were obtained by coupling N2,N2,N7-trimethylguanosine 5′-monophosphate P-imidazolide (16)19 with compounds 14 and 15, respectively, in the presence of excess ZnCl2 (Scheme 2). The role of ZnCl2 is to improve the solubility of reactants, activate the imidazole as a leaving group, and act as a template coordinating both nucleotide subunits that form the pyrophosphate bond.9 m3G cap analogs bearing a CH2 or NH substitution at the β/γ position of the 5′,5′-triphosphate bridge 4, 5 (Scheme 3) were obtained by coupling N2,N2,N7-trimethylguanosine 5′-methylene(bisphosphonate) (18)19 or P1-(N2,N2-dimethylguanosine-5′-yl)imidodiphosphate (19)19 with 12 (obtained by CDI-mediated activation) under similar conditions. β-Thio-modified m3G-cap analog 6 (6a – isomer D1 and 6b – isomer D2) was obtained by coupling 12 (obtained by Mukaiyama–Hashimoto method) with trimethylguanosine 5′-O-(β-thiodiphosphate) (20)19 leading to a mixture of two P-diastereoisomers of analog 6 (6a – isomer D1 and 6b – isomer D2)19 The α-thio-modified m3G-cap analogs 7a and b (7a – isomer D1 and 7b – isomer D2) were synthesized by coupling N2,N2,N7-trimethylguanosine-5′-O-(diphosphate) P-imidazolide (17)19 with 11. All of the synthesized m3G-caps 1–7 were isolated as triethylammonium salts using ion exchange chromatography with 40–60% yield. Further purification and, in the case of compounds 6 and 7, separation of P-diastereoisomers was performed by semi-preparative RP-HPLC yielding m3G caps 1–7 as ammonium salts. Final preparative yields ranged from 20 to 30%.
Stability of m3G-cap analogs in fetal bovine serum (FBS)
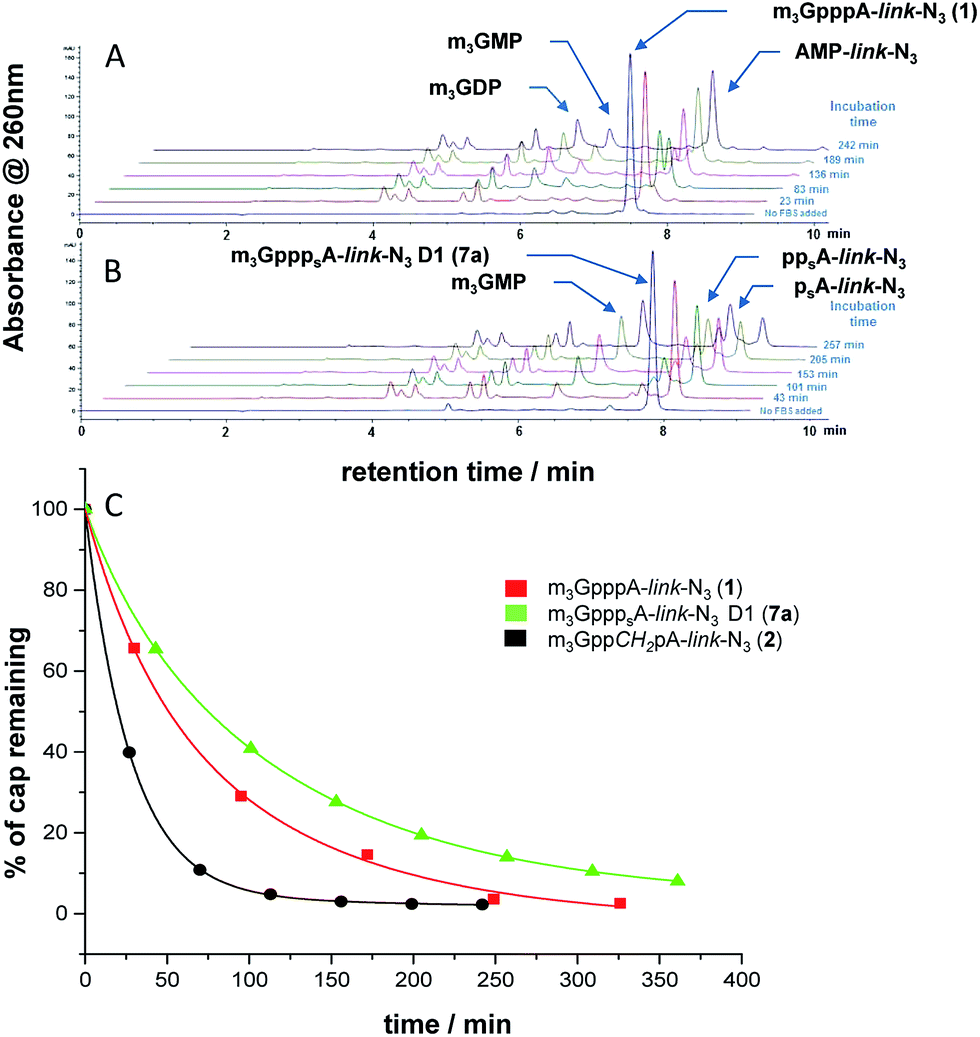
To determine the influence of various phosphate modifications on the general enzymatic susceptibility of the novel m3G-cap analogs, we studied their degradation in fetal bovine serum (FBS). FBS contains various nucleases capable of hydrolyzing pyrophosphate and phosphodiester bonds and has been previously employed to study the general stability of modified nucleotides and oligonucleotides under physiological conditions.37 Each m3G-cap analog was incubated in 2.5% FBS at 37 °C at a concentration of 10 μM. Samples taken at different time points were analyzed by RP-HPLC (Fig. 2).
 |
| | Fig. 2 Stability of m3G-cap analogs 1 (A) and 7a (B) in 2.5% FBS monitored by RP-HPLC. (C) Comparison of the stability of cap analogs 1, 2 and 7a based on the HPLC assay. | |
Based on the chromatographic integration, a percentage of the remaining substrate was estimated and plotted against time. An exponential decay model was fitted to experimental data to determine half-life values. (Fig. 2) The phosphate unmodified analog 1 (m3GpppAdo-link-N3) and m3GpppA without carbamoyl linker, had similar half-lives of 42.39 ± 3.49 and 40.25 ± 1.65 min, respectively, suggesting that carbamoyl substituent does not change the stability of cap analogs noticeably. Phosphate modifications influenced the stability of m3G-caps to various extent. First, cap analogs 2 (m3GppCH2pAdo-link-N3), 3 (m3GppNHpAdo-link-N3) and 4 (m3GpCH2ppAdo-link-N3), had all shorter half-lives (21.63 ± 1.31, 32.99 ± 8.19, 31.49 ± 2.62 min, respectively), indicating that bridging modifications at the α/β-phosphate decreased the stability in FBS. This is in contrast to previous finding on bridging methylene modifications in m3G cap with 2′-OMe substituents were both modified caps where more stable than the unmodified cap in 10% FBS in cell media.28 This suggests that the nature of the 2′-substituent does affect the relative cap stability. A stabilizing, effect was observed for β/γ imido modification 5 (m3GpNHppAdo-link-N3) with half-life of 58.81 ± 2.11 min. m3G caps bearing bridging modifications at β position 6a (m3GppspAdo-link-N3, D1) and 6b (m3GppspAdo-link-N3, D2) had half-life 52.03 ± 2.90 and 40.47 ± 1.10 min respectively showing a moderate effect of configuration on stability. Such effect is even more noticeable for compounds modified with phosphorothioate group at the α-position 7a (m3GpppsAdo-link-N3, D1) and 7b (m3GpppsAdo-link-N3, D2) with half-lives of 85.56 ± 5.51 and 28.96 ± 1.48 min respectively. This two-fold increased stability in FBS for the D1 isomer (7b) bearing sulfur modification at the α position indicate that the degradation of m3GpppAdo-link-N3 cap analogs in FBS occurs mainly through cleavage of the α,β-pyrophosphate bond which can also be concluded from our previous study where the α/β methylene modification showed greater stability in both FBS and cytosolic medium.28 However, lower stability of compounds 2 and 3 suggests that other cleavage pathways are also possible (Table 1).
Table 1 Stability of phosphate-modified m3G-cap analogs in FBS. The stability was tested for 10 μM cap analogs in 2.5% FBS diluted with PBS (pH 7.2)
| No |
Abbreviation |
Half-life [min] |
| |
m3GpppA |
40.25 ± 1.65 |
| 1 |
m3GpppA-link-N3 |
42.39 ± 3.49 |
| 2 |
m3GppCH2pA-link-N3 |
21.63 ± 1.31 |
| 3 |
m3GppNHpA-link-N3 |
32.99 ± 8.19 |
| 4 |
m3GpCH2ppA-link-N3 |
31.49 ± 2.62 |
| 5 |
m3GpNHppA-link-N3 |
58.81 ± 2.11 |
| 6a |
m3GppspA-link-N3 D1 |
52.03 ± 2.90 |
| 6b |
m3GppspA-link-N3 D2 |
40.47 ± 1.10 |
| 7a |
m3GpppsA-link-N3 D1 |
85.56 ± 5.51 |
| 7b |
m3GpppsA-link-N3 D2 |
28.96 ± 1.48 |
Conjugation with RNA using CuAAC
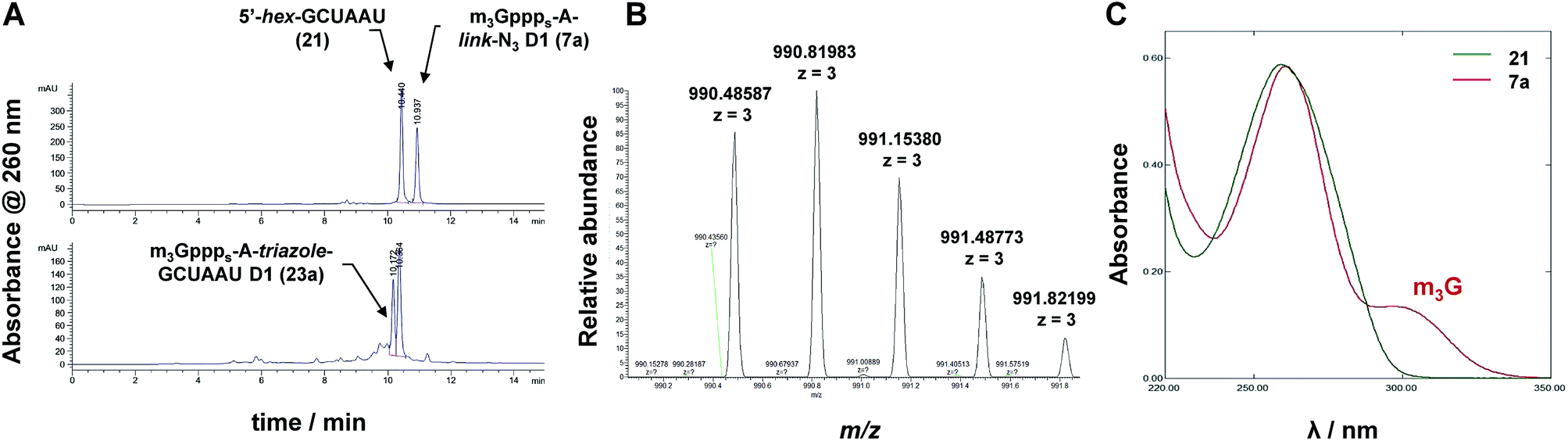
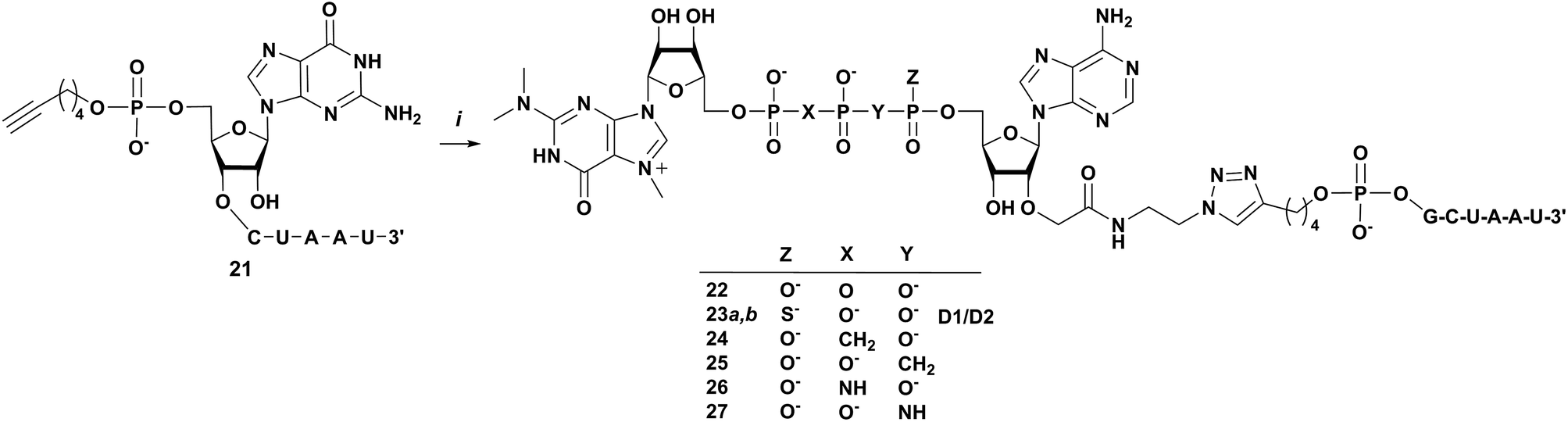
Next, we tested whether the novel analogs can be attached to 5′-functionalized short RNAs using CuAAC. The model RNA functionalized with 5′-hexynylphosphate was synthesized using standard phosphoroamidite solid phase synthesis protocol and using commercially available 5′-hexynyl phosphoramidate as the 5′-phosphitylating reagent. The 5′-hexynylphosphate-RNA (6 nt, 5′-hex-GCUAAU, 21) cleaved from the solid support, deprotected, and desalted by precipitation was pure enough to be used in “click” conjugation without additional purification. CuAAC reactions were carried out in H2O/DMSO mixture at 37 °C in the presence of catalytic amount of Cu–TBTA complex (0.05 equiv.) and excess sodium ascorbate at 37 °C. The respective m3G cap analog at 1 mM concentration was mixed with ∼2-fold excess of 5′-hex-GCUAAU and the reaction progress was monitored by RP-HPLC (Fig. 3). Six different clickable m3G-cap analogs 1–5, 7a and 7b were successfully conjugated to model short RNA using this procedure (Scheme 4). Due to relatively low reactant concentrations and a less reactive alkyne than in previous clicking of m3G caps,30 the reactions proceeded slowly, reaching 50–90% conversions (based on the amount of m3G cap-azide) within 3 to 5 days. The resulting m3G-capped conjugates were purified using analytical RP-HPLC, analyzed by HRMS (Table 2) and subjected to biochemical studies. In the case of m3G caps bearing a β-S modification 6a and 6b low yields of product after purification resulted in that amounts of isolated conjugate was insufficient for biological assays. This resulted from very slow reaction progress and overlapping HPLC signals of starting material and conjugate product.
 |
| | Fig. 3 Analysis of m3G cap analogs and their conjugates. (A) RP-HPLC profiles of the reaction mixture before (upper) and after 72 h (lower) click conjugation of m3G-cap 7a with 5′-hex-GCUAAU 21; (B) HRMS spectrum of the product m3GpppsA-triazole-GCUAAU D1 (23a); (C) UV spectra of oligonucleotide 21 and dinucleotide 7a. | |
 |
| | Scheme 4 Synthesis of m3GpppA-triazole-GCUAAU conjugates (22–27), (i) 1–3, 7a and 7b, CuSO4 × TBTA (1:1 complex, 5 mol%), sodium ascorbate, DMSO:H2O (1:1). | |
Table 2 HRMS data for m3G-cap-RNA conjugates (22–27)
| No |
Mol. formula |
m |
z |
Calc. m/z |
Found m/z |
| 21 |
C63H78N22O43P6 |
2016.3029 |
2 |
1008.1514 |
1008.1533 |
| 22 |
C90H116N36O61P9 |
2955.4720 |
3 |
985.1579 |
985.1605 |
| 23a |
C90H116N36O60P9S |
2971.4491 |
3 |
990.4836 |
990.4859 |
| 23b |
C90H116N36O60P9S |
2971.4491 |
3 |
990.4836 |
990.1499 |
| 24 |
C91H118N36O60P9 |
2953.4927 |
3 |
984.4981 |
984.5003 |
| 25 |
C91H118N36O60P9 |
2953.4927 |
3 |
984.4981 |
984.5000 |
| 26 |
C90H117N37O60P9 |
2955.4958 |
3 |
985.1658 |
985.8324 |
| 27 |
C90H117N37O60P9 |
2955.4958 |
3 |
985.1658 |
985.8324 |
Susceptibility of chemically capped short RNAs to degradation by hNUDT16
Next, we studied the susceptibility of m3G cap-RNA conjugates obtained by CuAAC to degradation by recombinant human decapping enzyme, hNUDT16.38 hNUDT16, belongs to the NUDIX family of phosphohydrolases,16 which contain metal binding acidic amino acids in the catalytic site and utilize substrates composed of a Nucleoside Diphosphate linked to a moiety X.39 hNUDT16 cleaves both m7G and m3G capped RNAs between the α and β phosphates to produce m7GDP or m3GDP and 5′-phosphorylated RNA.22 Here, we tested if the unmodified conjugate 22 bearing a triazole moiety (m3GpppA-triazole-RNA) is recognized by hNUDT16 and whether phosphate modifications would modulate the susceptibility to enzymatic degradation. The 5′-capped short RNAs obtained by click chemistry were incubated with hNUDT16 in vitro and the reaction products were analyzed on 20% denaturating polyacrylamide gel (Fig. 4) and the results of enzymatic degradation are summarized in Table 3. The susceptibility of the 5′-hex-RNA (21) to hNUDT16 was also tested. After resolving the gel was visualized by UV-shadowing at 254 nm (Fig. 4, left panel) to reveal both capped and decapped RNAs, and also using 302 nm wavelength UV lamp (Fig. 4, right panel) that allows to detect (oligo)nucleotides containing an m3G moiety (that have maximum absorption at 305 nm, as shown in Fig. 4, right). Since the capped products (5′-m3GpppA-triazole-RNA) migrate slower than uncapped RNA (hex-GCUAAU) the decapping reactions were observed by visualization at 254 nm (Fig. 4, left). Additionally, the m3G mononucleotides produced as a result of decapping, can be observed as fast migrating bands upon visualization at 302 nm. As shown in Fig. 4, m3GpppA-triazole-RNA (22) is cleaved by hNUDT16 releasing m3GDP (Fig. 4 right), indicating that the cleavage specificity between α and β phosphates is retained in the modified “click-capped” RNA. This suggests that the linker moiety between the m3G cap and the RNA does not disturb the recognition by hNUDT16. The O to NH substitution at the α/β produces RNA (m3GppNHpA-triazole-RNA, 27 and m3GppCH2pA-triazole-RNA, 25) that is resistant to decapping under conditions of our assay. In contrast, the m3G capped RNAs modified at the β/γ position (m3GpCH2ppA-triazole-RNA, 24 and m3GpNHppA-triazole-RNA 26) were efficiently cleaved by hNUDT16 to release products migrating slower than m3GDP and slightly faster than m3GMP, which we assigned as m3GpXp (where X is NH or CH2). The findings for 25, and 27 are in agreement with the regiospecificity of the enzyme observed for natural transcripts.20,21 It also fits well with the considerably higher stability of the α/β modification in cytosolic extract.28 However, surprisingly, transcripts modified at the α/β position with methylene group (m3GppCH2pA-triazole-RNA, 25) were also decapped to some extent, apparently through β/γ-pyrophosphate bond cleavage and release of m3GMP. This suggests that some modifications may direct the decapping to the β/γ-site, likely through a conformational change of the triphosphate bridge. That the decapping of conjugates 24 and 26 appears to be somewhat faster than for the unmodified 22 suggests also that the β/γ-modifications even activate decapping by hNUDT16. That observation was confirmed in an analogous assay carried out on unconjugated m3G dinucleotide cap analogs and higher concentration of hNUDT16: the m3GppNHpA-link-N3 appeared to be fully resistant to hNUDT16 degradation, whereas the m3GppCH2pA-link-N3 showed some susceptibility (1–7; ESI Fig. S1–S3†). The RNA capped with the m3GpppSA D2 23b is resistant to hNUDT16 decapping activity under the reaction conditions, but for the other diastereoisomer 23a, the degradation was observed. However, for its dinucleotide cap analogs 7a and 7b we observed higher stability of isomer D1. Most importantly, our results indicate that the O to NH substitution at the α,β-position is more efficient than the O to CH2 substitution for stabilizing m3G capped RNAs obtained by CuAAC, making this analog the best candidate for future studies on interaction with snurportin and intracellular transport.
 |
| | Fig. 4 Susceptibility of RNAs carrying variously modified m3G cap structures to degradation by hNUDT16. Short capped RNAs (m3Gcap-triazole-GCUAAU) were synthesized by “click chemistry” as described in Experimental section. Decapping reactions with hNUDT16 were incubated for 1 h at 37 °C in HCl–Tris pH 7.9. Thermally inactivated hNUDT16 was used in negative control reactions. Reaction products were separated on 20% denaturating polyacrylamide gel and visualized by UV 254 nm (left panel) and 302 nm (right panel). Visualization at 302 nm enables exclusive observation of m3G-containing (oligo)nucleotides due to distinct absorption and fluorescence properties of m3G (Fig. 3C). The capped RNAs that showed no difference for hNUDT16+ and hNUDT16− samples were assigned as resistant. | |
Table 3 Susceptibility to hNUDT16
| No |
Compound at the 5′ end of RNA |
Susceptibility to hNUDT16 |
| 22 |
m3GpppA-triazole |
Hydrolyzed |
| 23a |
m3GpppsA-triazole D1 |
Hydrolyzed |
| 23b |
m3GpppsA-triazole D2 |
Resistant |
| 24 |
m3GpCH2ppA-triazole |
Slowly hydrolyzed |
| 25 |
m3GppCH2pA-triazole |
Hydrolyzed |
| 26 |
m3GpNHppA-triazole |
Resistant |
| 27 |
m3GppNHpA-triazole |
Hydrolyzed |
Conclusions
In conclusion, we synthesized a set of m3G cap dinucleotides bearing an azido functionalized linker at the second nucleoside and various modifications within the 5′,5′-triphosphate bridge. The synthetic approach was based on phosphorimidazolide chemistry and enabled straightforward incorporation of either bridging (CH2, NH) or non-bridging (S) substitutions at different positions of the 5′,5′-triphosphate chain in combination with 2′-O-(N-(2-azidoethyl)carbamoyl)methyl group at the 2′-O-position of adenosine. The cap analogs were then successfully attached to short RNA using click chemistry approach. Despite the sterical hindrance introduced by the cap-RNA linker in m3G-triazole-RNAs, the capped RNAs were correctly recognized by hNUDT16 enzyme, as indicated by α/β cleavage of phosphate-unmodified m3G cap in dacapping assay. Interestingly, modifications at both the α/β and β/γ positions modulated the susceptibility to degradation by decapping enzyme, however, with distinct effects. Importantly, among studied compounds, only the bridging α/β O to NH substitution made the RNA and dinucleotide cap analogs resistant to hNUDT16, designating the compounds as most promising candidates for an efficient NLS signal. The bridging α/β O to CH2 substitution was also highly stabilizing but surprisingly, this analog was decapped slowly, presumably trough cleavage at the γ/β position. In contrast, the methylene or imido modifications at the β/γ position of triphosphate bridge appeared to increase the cleavage reaction rate. The influence of modification on non-specific degradation in fetal bovine serum was less pronounced. Nevertheless, the analogs containing the O to S substitution at the α-position showed increased stability. Therefore, the reported set of m3G cap analogs represents a useful toolbox for future studies aimed at assessing the importance of both type of degradations (specific or unspecific) under in vivo conditions. Moreover, further studies to assess the full potential of novel m3G cap analogs and their oligonucleotide conjugates, including affinity for snurportin and intracellular transport and stability are in progress.
Experimental
All chemicals and solvents were purchased from Sigma-Aldrich and used as received. For syntheses under anhydrous conditions solvents were additionally dried over 4 Å molecular sieves. POCl3 and PSCl3 were distilled prior to use. Silica gel column chromatography was performed on Kieselgel 60 Å (230–400 mesh, 40–63 μm) Thin Layer Chromatography (TLC) analysis was carried out on pre-coated Silica Gel 60 Å on aluminum foil with fluorescence indicator 254 nm (Sigma-Aldrich). N2,N2,N7-Trimethylguanosine 5′-monophosphate P-imidazolide (m3GMP-Im) (16) and N2,N2,N7-trimethylguanosine 5′-diphosphate P-imidazolide (m3GDP-Im, 17) were prepared as described by Honcharenko et al.28 N2,N2,N7-Trimethylguanosine 5′-methylene(bisphosphonate) (m3GpCH2p, 18), P1-(N2,N2-dimethylguanosine-5′-yl)imido-diphosphate (m3GpNHp, 19) and N2,N2,N7-trimethylguanosine 5′-(2-thiodiphosphate), (m3GDPβS, 20) were prepared as described recently19 Dichlorophosphorylphosphorimidoyl trichloride (PCl3NPOCl2) was prepared as described previously by Tomasz et al.33 with the exception that it was used in a liquid form for further reactions as problems with its crystallization occurred.33 2-Azidoethylamine31 and allyl bromoacetate31 were synthesized as previously described. Synthesized nucleotides were purified by ion-exchange chromatography on DEAE-Sephadex A-25 (HCO3−form) column. A column was loaded with reaction mixture and washed through with excess of water to remove metal (II) salt/EDTA complex. Then, the nucleotides were eluted using a linear gradient of triethylammonium bicarbonate (TEAB) in deionized water. After evaporation under reduced pressure with repeated additions of ethanol to decompose TEAB, compounds were isolated as triethylammonium (TEA) salts. Yields were calculated on the basis of either sample weight or (preferably) optical milliunits (opt. mu) of the product. Optical unit measurements were performed in 0.1 M phosphate buffer (pH 7 or pH 6 for m3G nucleotides) at 260 nm. Analytical HPLC was performed on Agilent Tech. Series 1200 using Supelcosil LC-18 T HPLC column (4.6 × 250 mm, flow rate 1.3 mL min−1) with a linear gradient 0–100% of methanol in 0.05 M ammonium acetate buffer (pH 5.9) in 15 min, UV-detection at 260 nm and fluorescence detection (excitation at 260 nm and detection at 370 nm). Semi preparative HPLC was performed on the same apparatus equipped with Discovery RP Amide C-16 HPLC column (25 cm × 21.2 mm, 5 μm, flow rate 5.0 mL min−1) with linear gradients of acetonitrile in 0.05 M ammonium acetate buffer (pH 5.9) and UV-detection at 260 nm. The structure and homogeneity of each final product was confirmed by RP HPLC, high resolution mass spectrometry HRMS (ESI−), 1H/31P NMR and FTIR spectroscopy. Intermediate products were characterized by low resolution MS (ESI−) or NMR. FTIR spectra were recorded on IRPrestige-21 spectrophotometer (Shimadzu Scientific Instruments) using ATR mode (4 cm−1 spectral resolution). Mass spectra were recorded on Thermo Scientific LTQ Orbitrap Velos (high resolution) and AB Sciex API 3200 (low resolution) spectrometers. 1H NMR and 31P spectra were recorded at 25 °C on a Varian UNITY-plus spectrometer at 399.94 MHz and 161.90 MHz, respectively. 1H NMR chemical shifts were reported to sodium 3-trimethylsilyl-[2,2,3,3-D4]-propionate (TSP) in D2O as an internal standard. 31P NMR chemical shifts were reported to 20% phosphorus acid in D2O as an external standard. Solvents and chemical reagents were purchased from Sigma-Aldrich and used without any pre-treatment unless otherwise stated. The raw NMR files were processed using ACD/NMR processor Academic Edition, version 12.01, Advanced Chemistry Development, Inc., Toronto, ON, Canada, http://www.acdlabs.com, 2014.
Chemical syntheses
2′-O-(N-(2-Azidoethyl)carbamoyl)methyladenosine (9)30. Adenosine (8) (500 mg, 1.87 mmol) was dried with vacuum pumping prior to use. The nucleoside was dissolved in dry DMF (50 ml) at r.t. Sodium hydroxide (NaH, 4 eq. 179.6 mg, 7.48 mmol) was added and after 10 min also allyl bromoacetate (2 eq., 669.5 mg, 3.74 mmol) stirring was continued for 1 h and then 2-azidoethylamine (3 eq., 258.2 mg, 5.61 mmol) was added and the reaction mixture was stirred for 24 h after which reaction mixture was concentrated on rotary evaporator. Crude product 9 was purified by column chromatography using a gradient of methanol in dichloromethane (from 0–10%). Fractions containing product were collected and concentrated yielding compound 9 as a yellow solid. Yield 9 (70–80%). Rt = 12.09 min; 1H NMR (400 MHz, D2O, 25 °C): δ [ppm] 8.35 (1H, s), 8.25 (1H, s), 6.22 (d, 1H, J = 6.5 Hz), 4.68 (dd, 1H, J = 6.2, 5.2 Hz), 4.59 (dd, 1H, J = 5.1, 3.1 Hz), 4.34 (q, 1H, J = 3.1 Hz), 4.24 and 4.11 (ABq, 2H, J = 15.22 Hz), 3.88 (ddd, 2H, J = 2.74, 3.49, 12.95 Hz), 3.36–3.24 (m, 4H); FTIR-ATR, ν [cm−1]: 2104.34 (N3), HRMS (ESI−) m/z 392.14309, calculated for C14H18N9O5; found 392.04209, [M − H]−.
2′-O-(N-(2-Azidoethyl)carbamoyl)methyladenosine 5′-monophosphate (10)30. 2′-O-(N-(2-Azidoethyl)carbamoyl)-methyladenosine (9) (200 mg, 0.51 mmol) dried overnight in a vaccum dessicator over P4O10 was suspended in trimethyl phosphate (10 ml) and place on ice-bath. After cooling to 0 °C freshly distilled POCl3 (3 eq., 1.53 mmol, 234.85 mg) was added into the mixture and reaction was stirred at 0 °C until the disappearance of the starting material as determined by RP-HPLC (usually 3–4 h). Then, reaction was stopped by addition of 0.7 M TEAB (pH 7) or 1 M NaHCO3aq until neutral pH was reached. The crude product was purified by DEAE-Sephadex and isolated as TEA salts. Yield 10: (4691 opt. mu, 255 mg, 75%) Rt = 9.34 min;1H NMR (400 MHz, D2O, 25 °C): δ [ppm] 8.57 (s, 1H), 8.38 (s, 1H), 6.34 (d, 1H J = 4.5 Hz), 4.69–4.65 (m, 2H), 4.48–4.44 (m, 1H), 4.30 and 4.21 (ABq, 2H, J = 15.44 Hz), 4.23–4.11 (m, 2H), 3.43–3.33 (m, 4H); 31P NMR (162 MHz, D2O, 25 °C): δ [ppm] 0.59; (s, 1P); FTIR-ATR, ν [cm−1]: 2104.34 (N3); HRMS (ESI−) m/z 472.10942, calculated for C14H19N9O8P; found 472.10938 [M − H]−.
2′-O-(N-(2-Azidoethyl)carbamoyl)methyladenosine 5′-monothiophosphate (11). 2′-O-(N-(2-Azidoethyl)carbamoyl)methyladenosine (9) (200 mg, 0.51 mmol) dried overnight in vaccum dessicator over P4O10 was suspended in trimethyl phosphate (10 ml) and place on ice-bath. After cooling to 0 °C freshly distilled PSCl3 (3 eq., 1.53 mmol, 259.33 mg) was added into the mixture and reaction was stirred at 0 °C until the disappearance of the starting material as determined by RP-HPLC (usually 2–3 h). Then, reaction was stopped by addition of 0.7 M TEAB (pH 7) or 1 M NaHCO3aq until neutral pH was reached. The crude product was purified by DEAE-Sephadex and isolated as TEA salts. Yield 11: (4043 opt. mu, 225 mg, 65%); Rt = 8.83 min; 1H NMR (400 MHz, D2O, 25 °C): δ [ppm] 8.66 (1H, s), 8.36 (s, 1H), 6.32 (d, 1H, J = 5.23 Hz), 4.7 (dd, 1H, J = 5.15, 3.85 Hz), 4.65 (dd, 1H, J = 5.5, 4.8 Hz), 4.50–4.46 (m, 1H), 4.21 and 4.30 (ABq, 2H, J = 15.44 Hz), 4.26–4.17 (m, 2H), 3.44–3.33 (m, 4H); 31P NMR (162 MHz, D2O, 25 °C): δ [ppm] 51.34; (s, 1P); FTIR-ATR, ν [cm−1] 2106.27 (N3); HRMS (ESI−) m/z 488.08658, calculated for C14H19N9O7PS; found 488.08654 [M − H]−.
2′-O-(N-(2-Azidoethyl)carbamoyl)methyladenosine 5′-phosphorimidazolide (12). 2′-O-(N-(2-Azidoethyl)carbamoyl)methyladenosine 5′-monophosphate (10) (TEA salt, 100 mg, 0.15 mmol), imidazole (102.12 mg, 1.5 mmol, 10 eq.), and 2,2′-dithiodipyridine (99.14 mg, 0.45 mmol, 3 eq.) were mixed in 1.5 mL anhydrous DMF. Triethylamine (33.06 μL, 45.53 mg, 0.45 mmol, 3 eq.) and triphenylphosphine (118.03 mg, 0.45 mmol, 3 eq.) were added, and the mixture was stirred for 3 h. Reaction progress was monitored by ESI-MS. The product was precipitated from reaction mixture with anhydrous NaClO4 (183.66 mg 1.5 mmol, 10 eq.) solution in dry acetone (15 mL). After cooling at 4 °C, the white precipitate was filtered, washed repeatedly with cold, dry acetone, and dried in vacuum dessicator over P4O10. Yield: 72 mg (93%): Rt = 13.48 min; FTIR-ATR, ν [cm−1]: 2108.20 (N3); HRMS (ESI−) m/z 522.13630, calculated for C17H21N11O7P; found 522.13762 [M − H]−.
2′-O-(N-(2-Azidoethyl)carbamoyl)methyladenosine 5′-diphosphate (13). 2′-O-(N-(2-Azidoethyl)carbamoyl)-methyladenosine 5′-phosphorimidazolide (12) (100 mg, 0.18 mmol) was dissolved in anhydrous DMF (2 mL), and tris(triethylammonium)-phosphate (100 mg, 0.26 mmol) was added, followed by addition of ZnCl2 (278.72 mg, 2.08 mmol), and the mixture was stirred at room temperature until the disappearance of the starting material as determined by RP-HPLC. Then, the reaction was stopped by addition of a solution of EDTA (607.36 mg, 2.08 mmol) in water (50 mL) and neutralized with 1 M NaHCO3. The crude product was purified by DEAE-Sephadex and isolated as TEA salts. Yield 13: (1406 opt. mu, 97 mg, 61%), Rt = 8.38 min; 1H NMR (400 MHz, D2O, 25 °C): δ [ppm] 8.54 (s, 1H), 8.30 (s, 1H), 6.30 (d, 1H, J = 5.5 Hz), 4.74–4.70 (m, 1H), 4.66–4.62 (m, 1H), 4.47–4.43 (m, 1H), 4.29 and 4.17 (ABq, 2H, J = 15.73 Hz), 4.26–4.23 (m, 2H), 3.40–3.30 (m, 4H); 31P NMR (162 MHz, D2O, 25 °C): δ [ppm] −10.19 (s, 1P α); −10.98 (s, 1P β) FTIR-ATR, ν [cm−1] 2106.27 (N3); HRMS (ESI−) m/z 552.07575, calculated for C14H20N9O11P2; found 552.07470 [M − H]−.
2′-O-(N-(2-Azidoethyl)carbamoyl)methyladenosine 5′-methylene(bisphosphonate) (14). 2′-O-(N-(2-Azidoethyl)-carbamoyl)methyladenosine (9) (200 mg, 0.51 mmol) dried overnight in vaccum dessicator over P4O10 was suspended in trimethyl phosphate (10 ml) and place on ice-bath. After cooling to 0 °C methylene bis(phosphonic dichloride) (CH2(POCl2)2, 3 eq., 1.53 mmol, 382.16 mg) was added into the mixture and reaction was stirred at 0 °C until the disappearance of the starting material as determined by RP-HPLC (usually 2–3 h). Then, reaction was stopped by addition of 0.7 M TEAB (pH 7) or 1 M NaHCO3aq until neutral pH was reached. The crude product was purified by DEAE-Sephadex and isolated as TEA salts. Yield: (3727 opt. mu, 257 mg, 59%), Rt = 7.77 min; 1H NMR (400 MHz, D2O, 25 °C): δ [ppm] 8.64 (s, 1H), 8.42 (s, 1H), 6.31 (d, 1H, J = 4.5 Hz), 4.71 (dd, 1H, J = 5.23, 4.98 Hz), 4.63 (dd, 1H, J = 4.90, 4.73 Hz), 4.46–4.42 (m, 1H), 4.31 and 4.26 (ABq, 2H, J = 15.44 Hz), 4.32–4.18 (m, 2H), 3.47–3.37 (m, 4H), 2.24 (t, 2H, J = 19.67 Hz); 31P NMR (162 MHz, D2O, 25 °C): δ [ppm] 18.64,(bs, P-α) 15.59; (m, P-β); FTIR-ATR, ν [cm−1] 2106.27 (N3); HRMS (ESI−) m/z, 550.09649 calculated for C15H22N9O10P2; found 550.09657 [M − H]−.
2′-O-(N-(2-Azidoethyl)carbamoyl)methyladenosine 5′-imidodiphosphate (15). 2′-O-(N-(2-Azidoethyl)carbamoyl)-methyladenosine (9) (200 mg, 0.51 mmol) dried overnight in vaccum dessicator over P4O10 was suspended in trimethyl phosphate (10 ml) and place on ice-bath. After cooling to 0 °C dichlorophosphoryl-phosphorimidoyl trichloride (Cl3PNP(O)Cl2, 3 eq., 1.53 mmol, 871.33 mg) was added into the mixture and reaction was stirred at 0 °C until the disappearance of the starting material as determined by RP-HPLC (usually 4–5 h). Then, reaction was stopped by addition of 0.7 M TEAB (pH 7) or 1 M NaHCO3aq until neutral pH was reached. The crude product was purified by DEAE-Sephadex and isolated as TEA salts. Yield: (3089 opt. mu, 213 mg, 49%), Rt = 8.26 min; 1H NMR (400 MHz, D2O, 25 °C): δ [ppm] 8.61 (s, 1H), 8.35 (s, 1H), 6.31 (d, 1H, J = 4.98 Hz), 4.73 (dd, 1H, J = 4.73, 4.48 Hz), 4.64 (t, 1H, J = 4.98 Hz), 4.48–4.44 (m, 1H), 4.32 and 4.23 (ABq, 2H, J = 15.44 Hz), 4.24–4.18 (m, 2H), 3.44–3.33 (m, 4H), 31P NMR (162 MHz, D2O, 25 °C): δ [ppm] 0.45,(s, P α), −1.07; (s, P β); FTIR-ATR, ν [cm−1] 2104.34 (N3); HRMS (ESI−) m/z, 551.09173 calculated for C14H21N10O10P2; found 551.09150 [M − H]−.
General procedure using m3G-imidazolides (Scheme 2)
m3GMP-Im, (16) or m3GDP-Im, (17) (1.5 eq.) and appropriate phosphates 13–15 or 11 were suspended in anhydrous DMF (1.0 mL) followed by addition of anhydrous ZnCl2 (38.88 mg, 10 eq., 0.29 mmol). The mixture was vigorously shaken until the reagents dissolved. The reaction progress was monitored by RP-HPLC. After completion, (24 h) appropriate amount of EDTA solution (Na2EDTA, 0.25 M, 1.16 mL, 0.29 mmol) was added to disassociate the nucleotide–metal complex, adjusted to pH 6 with solid NaHCO3, followed by purification by DEAE-Sephadex and isolated as TEA salts. Triethylammonium salts were then repurified by semipreparative RP-HPLC and after repeated freeze-drying, were isolated as ammonium salts.
General procedure using imidazolide 12 or in situ activation with CDI (Scheme 3)
In situ activation using CDI. AMP-link-N3 (10), (460 opt. mu, TEA salt, 25 mg, 0.037 mmol) was placed in a 10 mL microwave tube and suspended in anhydrous DMF (1.0 ml) followed by addition of carbodiimidazole (CDI, 5 eq., 30.11 mg, 0.185 mmol). The tube was heated for 20 min in the microwave oven using dynamic power mode (parameters: Pmax = 5 W and Tmax 40 ± 1 °C). After the reaction completion an excess of CDI was decomposed by addition of water (5 eq., 3.35 μl, 0.185 mmol).Then, to compound 12 or in situ activated compound 10 appropriate nucleotide 18, 19, or 20 followed by anhydrous ZnCl2 (75.65 mg, 10 eq., 0.55 mmol) were added. The reaction progress was monitored by RP-HPLC. After completion, (24 h) appropriate amount of EDTA solution (Na2EDTA, 0.25 M, 2.2 mL, 0.55 mmol) was added to disassociate the nucleotide–metal complex, adjusted to pH 6 with solid NaHCO3, followed by purification by DEAE-Sephadex and isolated as TEA salts. Triethylammonium salts were then repurified by semipreparative RP-HPLC and after repeated freeze-drying, were isolated as ammonium salts.
P1-(2,2,7-Trimethylguanosin-5′-yl)-P3-[2′-O-(N-(2-azidoethyl)-carbamoyl)methyl-adenosine-5′-yl]-triphosphate; m3GpppAdo-link-N3 (1)30. (469 opt. mu, 24 mg, 65%, TEA salt), (237 opt. mu, 9.7 mg, 51%, NH4+ salt), was obtained starting from (ADP-link-N3, TEA salt, 362 opt. mu, 25 mg, 0.029 mmol) and m3GMP-Im, (Na salt, 21.5 mg, 0.044 mmol, 1.5 eq.) following the general procedure 1. Rt = 7.54 min; 1H NMR (400 MHz, D2O, 25 °C): δ [ppm] 8.90 (s, 1H), 8.24 (s, 1H), 8.15 (s, 1H), 6.01 (d, 1H, J = 5.2 Hz), 5.82 (d, 1H, J = 3.2 Hz), 4.69–4.63 (m, 2H), 4.53–4.48 (m, 2H), 4.44 (bs, 1H), 4.39–4.19 (m, 7H), 4.09 (s, 3H); 3.31–3.21, (m, 4H), 3.06 (s, 6H), 31P NMR (162 MHz, D2O, 25 °C): δ [ppm]: −11.43 (m, 2P, P-α, P-γ), −23.05 (t, 1P, P-β, J = 17.2 Hz); FTIR-ATR, ν [cm−1] 2106.27 (N3); HRMS (ESI−) m/z, 939.17014 calculated for C27H38N14O18P3; found 939.16821 [M − 2H]−.
P1-(2,2,7-Trimethylguanosin-5′-yl)-P3-[2′-O-(N-(2-azidoethyl)-carbamoyl)methyladenosine-5′-yl]-1,2-methylene-triphosphate; m3GppCH2pAdo-link-N3 (2). (450 opt. mu, 23 mg, 63%, TEA salt); (149 opt. mu, 6.1 mg, 26%, NH4+ salt), was obtained starting from (pCH2p-Ado-linker-N3, TEA salt, 359 opt. mu, 25 mg, 0.029 mmol), and m3GMP-Im, (Na salt, 21.5 mg, 0.044 mmol, 1.5 eq.) following the general procedure 1. Rt = 9.38 min; 1H NMR (400 MHz, D2O, 25 °C): δ [ppm] 9.11 (s, 1H), 8.53 (s, 1H), 8.33 (s, 1H), 6.19 (d, 1H, J = 4.0 Hz), 5.98 (d, 1H, J = 3.5 Hz), 4.65–4.59 (m, 2H), 4.47–4.41 (m, 3H), 4.40–4.19 (m, 7H), 4.10 (s, 3H); 3.41–3.37, (m, 4H), 3.16 (s, 6H), 2.43 (td, 2H, J = 20.42, 3.49 Hz), 31P NMR (162 MHz, D2O, 25 °C): δ [ppm]: 17.08 (m, 1P, P-β), 8.74 (d, 1P, J = 16.11 Hz, P-γ), −10.79 (m, 1P, J = 23.19 Hz, P-α); FTIR-ATR, ν [cm−1] 2110.12 (N3); HRMS (ESI−) m/z, 937.19870 calculated for C28H40N14O17P3; found 937.18983 [M − 2H]−.
P1-(2,2,7-Trimethylguanosin-5′-yl)-P3-[2′-O-(N-(2-azidoethyl)-carbamoyl)methyladenosine-5′-yl]-1,2-imidotriphosphate; m3GppNHpAdo-link-N3 (3). (293 opt. mu, 15 mg, 40%, TEA salt) (81 opt. mu, 3.3 mg, 27%, NH4+ salt), was obtained starting from (pNHp-AMP-link-N3, TEA salt, 395 opt. mu, 25 mg, 0.029 mmol) and m3GMP-Im, (Na salt, 21.5 mg, 0.044 mmol, 1.5 eq.) following the general procedure 1. Rt = 9.33 min; 1H NMR (400 MHz, D2O, 25 °C): δ [ppm] 9.02 (s, 1H), 8.43 (s, 1H), 8.25 (s, 1H), 6.11 (d, 1H, J = 4.73 Hz), 5.92 (m, 1H), 4.61–4.55 (m, 2H), 4.45–4.33 (m, 4H), 4.28–4.08 (m, 6H), 4.05 (s, 3H); 3.34–3.29, (m, 4H), 3.12 (s, 6H), 31P NMR (162 MHz, D2O, 25 °C): δ [ppm]: −0.8 (m, 1P, P-α), −10.79 (m, 2P, P-β, P-γ); FTIR-ATR, ν [cm−1] 2108.20 (N3); HRMS (ESI−) m/z, 938.18612 calculated for C27H39N15O17P3; found 938.18545 [M − 2H]−.
P1-(2,2,7-Trimethylguanosin-5′-yl)-P3-[2′-O-(N-(2-azido-ethyl)carbamoyl)methyladenosine-5′-yl]-2,3-methylene-triphosphate; m3GpCH2ppAdo-link-N3 (4). (547 opt. mu, 28 mg, 60% TEA salt); (152 opt. mu, 6.2 mg, 27.7%, NH4+ salt) was obtained starting from m3GpCH2p, (18) (707 opt. mu, TEA salt, 43.56 mg, 0.056 mmol, 1.5 eq.) and AMP-link-N3, (10) (460 opt. mu, TEA salt, 25 mg, 0.037 mmol) following the general procedure 2. Rt = 9.39 min;1H NMR (400 MHz, D2O, 25 °C): δ [ppm] 9.29 (s, 1H), 8.55 (s, 1H), 8.37 (s, 1H), 6.23 (d, 1H, J = 4.5 Hz), 6.03 (d, 1H, J = 3.2 Hz), 4.69–4.63 (m, 2H), 4.53–4.48 (m, 2H), 4.44 (bs, 1H), 4.39–4.19 (m, 7H), 4.09 (s, 3H); 3.43–3.36, (4H, m), 3.18 (s, 6H), 2.43 (t, 2H, J = 20.17 Hz), 31P NMR (162 MHz, D2O, 25 °C): δ [ppm]: 17.29 (m, 1P, P-β), 7.76 (m, 1P, P-α), −10.93 (m, 1P, P-γ); FTIR-ATR, ν [cm−1] 2101.34 (N3); HRMS (ESI−) m/z, 937.19087 calculated for C28H40N14O17P3; found 937.19045 [M − 2H]−.
P1-(2,2,7-trimethylguanosin-5′-yl)-P3-[2′-O-(N-(2-azidoethyl)-carbamoyl)methyladeno-sine-5′-yl]-2,3-imidotriphosphate; m3GpNHppAdo-link-N3 (5). (371 opt. mu, 19 mg, 40%, TEA salt); (132 opt. mu, 5.4 mg, 35%, NH4+ salt), was obtained starting from m3GpNHp, (19) (707 opt. mu, TEA salt, 43.62 mg, 0.056 mmol, 1.5 eq.) and AMP-link-N3, (10) (460 opt. mu, TEA salt, 25 mg, 0.037 mmol) following the general procedure 2. Rt = 9.73 min; 1H NMR (400 MHz, D2O, 25 °C): δ [ppm] 9.13 (s, 1H), 8.36 (s, 1H), 8.24 (s, 1H), 6.11 (d, 1H, J = 4.73 Hz), 5.91 (m, 1H), 4.62–4.55 (bs, 2H), 4.48–4.10 (m, 10H), 4.08 (s, 3H); 3.39–3.31, (m, 4H), 3.13 (s, 6H), 31P NMR (162 MHz, D2O, 25 °C): δ [ppm]: −0.87 (m, 1P, P-α), −11.00 (m, 2P, P-β, P-γ); FTIR-ATR, ν [cm−1] 2108.20 (N3); HRMS (ESI−) m/z, 938.18612 calculated for C27H39N15O17P3; found 938.18215 [M − 2H]−.
P1-(2,2,7-Trimethylguanosin-5′-yl)-P3-[2′-O-(N-(2-azidoethyl)carbamoyl)methyladenosine-5′-yl]-2-thio-triphosphate; m3GppspAdo-link-N3 (6a and 6b). (480 opt. mu, 24.9 mg, yield 53%, mixture of diastereoisomers as a triethylammonium salt); D1 (212 opt. mu, 8.8 mg, 44%, NH4+ salt) and D2 (159 opt. mu, 6.6 mg, 33%, NH4+ salt) were obtained starting from (Im AMP-link-N3, Na salt, 30.5 mg, 0.056 mmol, 1.5 eq.) and m3GDP-βS, (TEA salt, 30.0 mg, 0.037 mmol) following the general procedure 2. D1: 1H NMR (400 MHz, D2O, 25 °C): δ [ppm] 8.33 (s, 1H), 8.18 (s, 1H), 6.07 (d, 1H, J = 5.48 Hz), 5.89 (d, J = 3.52 Hz), 4.59 (dd, 1H, J = 4.70, 4.30 Hz), 4.52 (dd, 1H, J = 4.30, 3.91 Hz), 4.45–4.39 (m, 4H), 4.37–4.22 (m, 4H), 4.17 and 4.05 (ABq, 2H, J = 15.26 Hz), 4.02 (s, 3H), 3.31–3.32, (m, 4H), 3.09 (s, 6H), 31P NMR (162 MHz, D2O, 25 °C): δ [ppm]: 29.89 (m, 1P, P-β), −12.46 (t, 2P, J = 27.88, P-α, P-γ); FTIR-ATR, ν [cm−1] 2104.34 (N3); HRMS (ESI−) m/z, 955.14729 calculated for C27H38N14O17P3S; found 955.14670 [M − 2H]−. D2: 1H NMR (400 MHz, D2O, 25 °C): δ [ppm] 8.30 (s, 1H), 8.16 (s, 1H), 6.04 (d, 1H, J = 5.48 Hz), 5.84 (d, J = 3.91 Hz), 4.57 (dd, 1H, J = 4.30, 4.70 Hz), 4.52 (t, 1H, J = 4.30), 4.43–4.33 (m, 4H), 4.29–4.21 (m, 4H), 4.17 and 4.05 (ABq, 2H, J = 15.26 Hz), 4.03 (s, 3H), 3.32–3.23, (m, 4H), 3.08 (s, 6H), 31P NMR (162 MHz, D2O, 25 °C): δ [ppm]: 29.50 (t, 1P, J = 24.94 Hz, P-β), −12.71 (dd, 2P, J = 24.94 Hz, P-α, P-γ); FTIR-ATR, ν [cm−1] 2106.27 (N3); HRMS (ESI−) m/z, 955.14729 calculated for C27H38N14O17P3S; found 955.14711 [M − 2H]−.
P1-(2,2,7-Trimethylguanosin-5′-yl)-P3-[2′-O-(N-(2-azidoethyl)carbamoyl)methyladenosine-5′-yl]-3-thiotriphosphate; m3GpppsAdo-link-N3 (7a and 7b). (542 opt. mu, 28.1 mg, yield 60%, mixture of diastereoisomers as a triethylammonium salt); D1 (152 opt. mu, 6.3 mg, 28.0%, NH4+ salt) and D2 (111 opt. mu, 4.6 mg, 20.5%, NH4+ salt) were obtained starting from (AMPS-link-N3, TEA salt, 446 opt. mu, 25.0 mg, 0.036 mmol) and m3GDP-Im, (Na salt, 31.0 mg, 0.054 mmol, 1.5 eq.) following the general procedure 1. D1: 1H NMR (400 MHz, D2O, 25 °C): δ [ppm] 9.01 (s, 1H), 8.43 (s, 1H), 8.24 (s, 1H), 6.12 (d, 1H, J = 5.45 Hz), 5.95 (d, J = 3.44 Hz), 4.64, (t, 1H, J = 4.48), 4.58 (t, 1H, J = 4.23 Hz), 4.47–4.27 (m, 8H), 4.23 and 4.11 (ABq, 2H, J = 15.44 Hz), 4.08 (s, 3H); 3.40–3.28, (m, 4H), 3.14 (s, 6H), 31P NMR (162 MHz, D2O, 25 °C): δ [ppm]: 43.67 (d, 1P, J = 21.63 Hz, P-α), −11.45 (d, 1P, J = 16.60 Hz, P-γ), −23.87, −24.01 (2d, 1P, J = 20.75, 21.97 Hz, P-β); FTIR-ATR, ν [cm−1] 2106.27 (N3); HRMS (ESI−) m/z, 955.14729 calculated for C27H38N14O17P3S; found 955.14762 [M − 2H]−. D2: 1H NMR (400 MHz, D2O, 25 °C): δ [ppm] 9.00 (s, 1H), 8.46 (s, 1H), 8.24 (s, 1H), 6.12 (d, 1H, J = 5.23 Hz), 5.92 (d, J = 3.49 Hz), 4.60, (dd, 1H, J = 4.48, 4.73 Hz), 4.55 (dd, 1H, J = 4.23, 3.98 Hz), 4.53–4.35 (m, 6H), 4.34–4.23 (m, 2H), 4.22 and 4.12 (ABq 2H, J = 15.69 Hz), 4.07 (s, 3H); 3.39–3.28, (m, 4H), 3.13 (s, 6H); 31P NMR (162 MHz, D2O, 25 °C): δ [ppm]: 43.95 (d, 1P, J = 23.68 Hz, P-α), −11.47 (d, 1P, J = 16.36 Hz, P-β), −23.76, −23.89 (2d, 1P, J = 22.95, 20.02 Hz, P-γ) FTIR-ATR, v [cm−1] 2108.20 (N3); HRMS (ESI−) m/z, 955.14729 calculated for C27H38N14O17P3S; found 955.14770 [M − 2H]−.
Synthesis of 5′-hexynyl-GCUAAU (21)
RNA synthesis was performed on AKTA Oligopilot plus 10 synthesizer (GE Healthcare) on a 5 μmol scale using a 1.2 mL column filled with commercially available solid support – Custom Primer Support™ Ribo U 40 from GE Healthcare (Primer support 200 at 39 μmol g−1). The detritilation reagent was 3% (v/v) dichloroacetic acid in toluene (Novabiochem® Deblocking Reagent from Merck). In the coupling step, the 20 equivalents of appropriate acetyl base protected 2′-O-TBDMS phosphoramidate (rAAc, rCAc, rGAc, rU from ChemGenes) and 0.30 M 5-(benzylthio)-1H-tetrazole in acetonitrile (Novabiochem® Activator Reagent from Merck) were recirculated through the column for 15 minutes. The last coupling was performed in the same conditions using hexynyl phosphoramidate (ChemGenes), yet the detritilation step was omitted due to lack of DMT-tag in this phosphoramidate. As an oxidizing reagent, 0.05 M solution of iodine in pyridine (Novabiochem® Oxidizing Reagent from Merck) was employed. Finally, RNA still on the solid support was treated with 20% (v/v) diethylamine in acetonitrile (Novabiochem® Merck) to remove 2-cyanoethyl protecting groups. Solid support was washed with acetonitrile, dried with air and transferred to a 50 mL plastic tube. Functionalized oligonucleotide was released from solid support and base protecting groups were removed using 5 mL of 1:1 mixture of 25% aqueous ammonia and 40% aqueous methylamine (AMA). After 60 minutes of incubation at 60 °C the solution was filtered off and the support was washed with 50% ethanol, twice. Combined filtrates were evaporated, lyophilized from water and re-dissolved in 500 μL of DMSO. 625 μL of triethylammonium trihydrofluoride was added and the sample was shaken and incubated at 65 °C for 2.5 hours. Then, 125 μL of 3 M sodium acetate was added followed by 5 mL of 1-butanol and the mixture was placed at −80 °C for 30 minutes. Precipitate of oligonucleotide sodium salt was centrifuged, lyophilized from water and used for click reaction without additional purification.
Conjugation of the “clickable” m3G-cap constructs (1–7) to the 5′-hexynyl-GCUAAU (21)
5′-hexynyl-GCUAAU (1.0 OD, 28 μg, 14 nmol, 3 eq.), dissolved in (in 15.5 μL of H2O:DMSO (1:1) was mixed with m3G-cap 1–7 (0.1 OD, 4.5 μg, 4.6 nmol, 1.5 ml, 3 mM). Then Cu–TBTA complex (1.0 ml, 10 mM in 55 vol% DMSO to final concentration of 0.5 mM) and sodium ascorbate (2.0 ml, 5 mM in H2O to final concentration of 0.5 mM). The solution was gently agitated on a vortex, centrifuged and thermostated at 37 °C for few days (3–5). Purification was done by analytical RP-HPLC using a linear gradient of buffer B in A from 0 to 100% B in 15 min, detection at 260 nm.
FBS stability assay
For the serum stability assay, 100 μM stock solutions of compounds (18–27) in MilliQ water were prepared. Then, 100 μL of respective stock solution, 890 μL of PBS buffer (pH 7.2, 140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4) and 10 μL of FBS were mixed in 1.5 mL HPLC vials, so each solution contained 2.5% of FBS and the final compound concentration was 10 μM. Immediately after FBS addition, the reaction mixtures were set up in a thermostated compartment maintained at 37 °C, and 100 μL injections were made with an automated sample injector at various time points. Usually, a single sample set covered unmodified cap analog 1 and a pair of α/β and β/γ isomers, or D1 and D2 isomers bearing the same modification, so that each compound could be injected at 8 different time points at 60 min intervals. Afterwards, each assay column was washed with methanol for at least 1 h before the next analysis. Based on the integration of the remaining cap analog and resultant products, substrate conversion for each compound was estimated using the equation: ∑(AUCp/ε)/∑[(AUCp/ε) + AUCs/ε]ε where AUC(p,s) – area under curve of products (p) and starting material (s), ε – molar extinction coefficient. Extinction coefficient values used for calculations were as follows: for trimethylated products ε = 12640 M−1 cm−1, for adenosine analogs ε = 12400 M−1 cm−1 and for the m3G-cap analogs ε = 24250 M−1 cm−1. The obtained data were analyzed using OriginPro software v. 9.1.
Protein expression and purification
Human Nudt16 protein (hNudt16, 1-199 aa, MW 22 kDa) was expressed in E. coli Rosetta2 (DE3) as a C-terminally His-tagged protein using a pET16b_Nudt16 vector, where the coding sequence of Nudt16 was cloned at the NcoI-BamHI restriction sites (verified further by sequencing). A sequence encoding four additional histidines was introduced just after the two terminal histidines of Nudt16 what resulted in a 6×His C-terminal His-tag. Expression was induced at OD600 ∼ 0.6 with 0.2 mM IPTG and after 4 hours incubation at 37 °C bacterial cells were pelleted and washed in PBS buffer. Subsequently, the pelleted cells were resuspended in a lysis buffer (20 mM HEPES-KOH pH 8.0, 300 mM NaCl, 300 mM urea, 10 mM imidazole, 10% glycerol, 1% Triton 100) supplemented with lysozyme, incubated on ice for 30 minutes and then disrupted by sonication. After centrifugation (17500g for 30 min), the supernatant was added to NTA-agarose equilibrated with 20 mM HEPES buffer (20 mM HEPES-KOH pH 8.0, 300 mM NaCl, 10% glycerol and 5 mM imidazole) and incubated for 1 hour in a cold room with gentle stirring. Unbound proteins were removed by washing with 20 mM Tris–HCl pH 8.0, 300 mM NaCl (10 × 2 mL). The His-tagged hNudt16 protein was eluted with the increasing concentrations of imidazole (20 mM to 300 mM) in wash buffer (20 mM Tris–HCl pH 8.0, 300 mM NaCl). The collected fractions of pure hNudt16 protein were dialyzed against 50 mM Tris–HCl pH 8.0, 150 mM KCl and 20% glycerol. Aliquots of purified hNudt16 were supplemented with 1 mM DTT, frozen in liquid nitrogen and stored at −80 °C.
Analysis of susceptibility of modified m3G dinucleotides and capped ribooligonucleotides towards hNUDT16 decapping
Short m3G-capped RNAs (m3G-cap-GCUAAU) containing various cap structures (24–29) were chemically synthesized by using click chemistry approach. Decapping reactions with hNUDT16 of clicked ribooligonucleotides and cap dinucleotides were performed in a final volume of 10 μL in 40 mM Tris–HCl buffer (pH 7.9, 6 mM MgCl2, 10 mM NaCl, 10 mM DTT and 2 mM spermidine) for 1 hour at 30 °C. Final concentration of hNudt16 in the reaction was 0.35 μM. To negative control reactions the same concentration of thermally inactivated hNUDT16 enzyme (for 8 minutes at 100 °C) was added. In the case of capped hex-oligo-RNA, around 500 pmoles of each substrate was present in the reaction (concentration of capped hex-oligo-RNA was estimated using PicoDrop spectrophotometer set up to oligo mode). In the case of dinucleotide analogs each substrate was added to ∼0.3 mM concentration. Reactions were stopped by addition of equal volume of formazol and incubation for 5 min at 55 °C. Reaction products were separated on 20% denaturating polyacrylamide gel with 7 M urea, and visualized by two methods: first by UV-shadowing using 254 nm UV hand lamp and digital camera, and second – using ChemiDoc (Biorad) gel visualization system (with 302 nm UV lamp) set up to ethidium bromide mode.
Acknowledgements
This project was supported by a grants from the Polish National Center of Research and Development (02/EuroNanoMed/2011), The National Science Centre (Poland, UMO-2012/05/E/ST5/03893, UMO-2013/08/A/NZ1/00866) and the Swedish Research Council.
References
- J. D. Thomas, R. C. Conrad and T. Blumenthal, Cell, 1988, 54, 533–539 CrossRef CAS PubMed.
- R. F. Liou and T. Blumenthal, Mol. Cell. Biol., 1990, 10, 1764–1768 CrossRef CAS PubMed.
- S. Hausmann and S. Shuman, J. Biol. Chem., 2005, 280, 4021–4024 CrossRef CAS PubMed.
- J. Mouaikel, C. Verheggen, E. Bertrand, J. Tazi and R. Bordonne, Mol. Cell, 2002, 9, 891–901 CrossRef CAS PubMed.
- A. G. Matera, R. M. Terns and M. P. Terns, Nat. Rev. Mol. Cell Biol., 2007, 8, 209–220 CrossRef CAS PubMed.
- S. Kitao, A. Segref, J. Kast, M. Wilm, I. W. Mattaj and M. Ohno, Mol. Cell. Biol., 2008, 28, 487–497 CrossRef CAS PubMed.
- J. Huber, U. Cronshagen, M. Kadokura, C. Marshallsay, T. Wada, M. Sekine and R. Luhrmann, EMBO J., 1998, 17, 4114–4126 CrossRef CAS PubMed.
- J. Hamm, E. Darzynkiewicz, S. M. Tahara and I. W. Mattaj, Cell, 1990, 62, 569–577 CrossRef CAS PubMed.
- M. Kadokura, T. Wada, C. Urashima and M. Sekine, Tetrahedron Lett., 1997, 38, 8359–8362 CrossRef CAS.
- M. Jankowska-Anyszka, K. Piecyk and J. Samonina-Kosicka, Org. Biomol. Chem., 2011, 9, 5564–5572 CAS.
- A. Ohkubo, Y. Kondo, M. Suzuki, H. Kobayashi, T. Kanamori, Y. Masaki, K. Seio, K. Nagai and M. Sekine, Org. Lett., 2013, 15, 4386–4389 CrossRef CAS PubMed.
- P. M. Moreno, M. Wenska, K. E. Lundin, O. Wrange, R. Stromberg and C. I. Smith, Nucleic Acids Res., 2009, 37, 1925–1935 CrossRef CAS PubMed.
- J. C. van Deutekom, A. A. Janson, I. B. Ginjaar, W. S. Frankhuizen, A. Aartsma-Rus, M. Bremmer-Bout, J. T. den Dunnen, K. Koop, A. J. van der Kooi, N. M. Goemans, S. J. de Kimpe, P. F. Ekhart, E. H. Venneker, G. J. Platenburg, J. J. Verschuuren and G. J. van Ommen, N. Engl. J. Med., 2007, 357, 2677–2686 CrossRef CAS PubMed.
- A. H. Burghes and V. L. McGovern, Genes Dev., 2010, 24, 1574–1579 CrossRef CAS PubMed.
- A. G. L. Douglas and M. J. A. Wood, Mol. Cell. Neurosci., 2013, 56, 169–185 CrossRef CAS PubMed.
- G. Lu, J. Zhang, Y. Li, Z. Li, N. Zhang, X. Xu, T. Wang, Z. Guan, G. F. Gao and J. Yan, Protein Cell, 2011, 2, 64–73 CrossRef CAS PubMed.
- S. Shukla and R. Parker, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E3277–E3286 CrossRef CAS PubMed.
- J. Jemielity, J. Kowalska, A. M. Rydzik and E. Darzynkiewicz, New J. Chem., 2010, 34, 829–844 RSC.
- M. Zytek, J. Kowalska, M. Lukaszewicz, B. A. Wojtczak, J. Zuberek, A. Ferenc-Mrozek, E. Darzynkiewicz, A. Niedzwiecka and J. Jemielity, Org. Biomol. Chem., 2014, 12, 9184–9199 CAS.
- E. Grudzien-Nogalska, J. Jemielity, J. Kowalska, E. Darzynkiewicz and R. E. Rhoads, RNA, 2007, 13, 1745–1755 CrossRef CAS PubMed.
- E. Grudzien, M. Kalek, J. Jemielity, E. Darzynkiewicz and R. E. Rhoads, J. Biol. Chem., 2006, 281, 1857–1867 CrossRef CAS PubMed.
- W. Su, S. Slepenkov, E. Grudzien-Nogalska, J. Kowalska, M. Kulis, J. Zuberek, M. Lukaszewicz, E. Darzynkiewicz, J. Jemielity and R. E. Rhoads, RNA, 2011, 17, 978–988 CrossRef CAS PubMed.
- M. Ziemniak, M. Strenkowska, J. Kowalska and J. Jemielity, Future Med. Chem., 2013, 5, 1141–1172 CrossRef CAS PubMed.
- J. Kowalska, M. Lukaszewicz, J. Zuberek, M. Ziemniak, E. Darzynkiewicz and J. Jemielity, Bioorg. Med. Chem. Lett., 2009, 19, 1921–1925 CrossRef CAS PubMed.
- A. M. Rydzik, M. Kulis, M. Lukaszewicz, J. Kowalska, J. Zuberek, Z. M. Darzynkiewicz, E. Darzynkiewicz and J. Jemielity, Bioorg. Med. Chem., 2012, 20, 1699–1710 CrossRef CAS PubMed.
- E. Grudzien, M. Kalek, J. Jemielity, E. Darzynkiewicz and R. E. Rhoads, J. Biol. Chem., 2006, 281, 1857–1867 CrossRef CAS PubMed.
- M. Ziemniak, M. Strenkowska, J. Kowalska and J. Jemielity, Future Med. Chem., 2013, 5, 1141–1172 CrossRef CAS PubMed.
- M. Honcharenko, M. Zytek, B. Bestas, P. Moreno, J. Jemielity, E. Darzynkiewicz, C. I. Smith and R. Strömberg, Bioorg. Med. Chem., 2013, 21, 7921–7928 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed. Engl., 2001, 40, 2004–2021 CrossRef CAS.
- M. Honcharenko, J. Romanowska, M. Alvira, M. Jezowska, M. Kjellgren, C. I. Smith and R. Stromberg, RSC Adv., 2012, 2, 12949–12962 RSC.
- M. Wenska, S. Milton and R. Strömberg, Protocol Exchange, 2010 DOI:10.1038/nprot.2010.93.
- M. Yoshikawa, T. Kato and T. Takenishi, Tetrahedron Lett., 1967, 8, 5065–5068 CrossRef.
- J. Tomasz, M. M. Vaghefi, P. C. Ratsep, R. C. Willis and R. K. Robins, Nucleic Acids Res., 1988, 16, 8645–8664 CrossRef CAS PubMed.
- M. Warminski, J. Kowalska, J. Buck, J. Zuberek, M. Lukaszewicz, C. Nicola, A. N. Kuhn, U. Sahin, E. Darzynkiewicz and J. Jemielity, Bioorg. Med. Chem. Lett., 2013, 23, 3753–3758 CrossRef CAS PubMed.
- M. Strenkowska, P. Wanat, M. Ziemniak, J. Jemielity and J. Kowalska, Org. Lett., 2012, 14, 4782–4785 CrossRef CAS PubMed.
- T. Mukaiyama and M. Hashimoto, J. Am. Chem. Soc., 1972, 94, 8528–8532 CrossRef CAS PubMed.
- J. W. Conway, C. K. McLaughlin, K. J. Castor and H. Sleiman, Chem. Commun., 2013, 49, 1172–1174 RSC.
- M.-G. Song, S. Bail and M. Kiledjian, RNA, 2013, 19, 390–399 CrossRef CAS PubMed.
- M. J. Bessman, D. N. Frick and S. F. O'Handley, J. Biol. Chem., 1996, 271, 25059–25062 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra25684d |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b,
Maciej Lukaszewiczb,
Malgorzata Honcharenkoc,
C. I. Edvard Smithd,
Roger Strömbergc,
Edward Darzynkiewicza and
Jacek Jemielity
b,
Maciej Lukaszewiczb,
Malgorzata Honcharenkoc,
C. I. Edvard Smithd,
Roger Strömbergc,
Edward Darzynkiewicza and
Jacek Jemielity