
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stepwise synthesis of mixed-metal assemblies using pre-formed Ru(II) ‘complex ligands’ as building blocks†
Alexander J.
Metherell
and
Michael D.
Ward
*
Department of Chemistry, University of Sheffield, Sheffield S3 7HF, UK. E-mail: m.d.ward@sheffield.ac.uk
First published on 14th January 2016
Abstract
Two families of heteronuclear coordination complexes have been prepared in a stepwise manner using pre-formed, kinetically inert [RuL3]2+ building blocks, in which L is a bis-bidentate bridging ligand with two pyrazole–pyridyl termini, coordinated at one end to the Ru(II) centre. These pre-formed ‘complex ligands’ – with three pendant binding sites – react with additional labile transition metal dications to complete the stepwise assembly of mixed-metal arrays in which labile [Co(II)/Cd(II)] or inert [Ru(II)] ions strictly alternate around the framework. When L = the thiophene-2,5-diyl spaced ligand Lth, the complex [Ru(Lth)3]2+ is formed in the expected 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mer:fac ratio: reaction with labile Co(II) or Cd(II) ions completes formation of a heteronuclear square [Ru2Co2(Lth)6]8+ or one-dimensional coordination polymer {[CdRu(Lth)3]4+}∞, respectively. In these only the mer isomer of [Ru(Lth)3]2+ is selected by the self-assembly process, whereas the fac isomer is not used. When L = a 1,3-benzene-diyl spaced ligand (Lph), the complex ligand [Ru(Lph)3]2+ formed in the initial step is enriched in mer isomer (80–87% mer, depending on reaction conditions). Two quite different products were isolated from reaction of [Ru(Lph)3]2+ with Co(II) depending on the conditions. These are the rectangular, hexanuclear ‘open-book’ array [Ru3Co3(Lph)9]12+ which contains a 2:1 proportion of fac/mer Ru(II) metal centres; and the octanuclear cubic [Ru4Co4(Lph)12{Na(BF4)4}]13+ cage which is a new structural type containing all mer Ru(II) vertices and all fac Co(II) vertices. The cavity of this cubic cage contains a tetrahedral array of fluoroborate anions which in turn coordinate to a central Na(I) ion – an unusual example of a metal complex [Na(BF4)4]3− acting as the guest inside the cage-like metal complex [Ru4Co4(Lph)12]16+.
:1 mer:fac ratio: reaction with labile Co(II) or Cd(II) ions completes formation of a heteronuclear square [Ru2Co2(Lth)6]8+ or one-dimensional coordination polymer {[CdRu(Lth)3]4+}∞, respectively. In these only the mer isomer of [Ru(Lth)3]2+ is selected by the self-assembly process, whereas the fac isomer is not used. When L = a 1,3-benzene-diyl spaced ligand (Lph), the complex ligand [Ru(Lph)3]2+ formed in the initial step is enriched in mer isomer (80–87% mer, depending on reaction conditions). Two quite different products were isolated from reaction of [Ru(Lph)3]2+ with Co(II) depending on the conditions. These are the rectangular, hexanuclear ‘open-book’ array [Ru3Co3(Lph)9]12+ which contains a 2:1 proportion of fac/mer Ru(II) metal centres; and the octanuclear cubic [Ru4Co4(Lph)12{Na(BF4)4}]13+ cage which is a new structural type containing all mer Ru(II) vertices and all fac Co(II) vertices. The cavity of this cubic cage contains a tetrahedral array of fluoroborate anions which in turn coordinate to a central Na(I) ion – an unusual example of a metal complex [Na(BF4)4]3− acting as the guest inside the cage-like metal complex [Ru4Co4(Lph)12]16+.
Introduction
Polynuclear coordination cages – hollow, pseudo-spherical metal/ligand capsules – are a field of major importance within supramolecular chemistry.1 Originally interest in them arose because of the possibility of making elaborate new structures from simple components by self-assembly methods. With this starting to become a mature field, the focus is now shifting towards the functional behaviour that can arise when guests bind in the central cavity.2–5 The vast majority of coordination cages – even those with very elaborate structures – are based on just two types of component, i.e. one type of metal ion and one type of bridging ligand.1 Whilst this is not important if the cage is acting simply as a container with a central cavity having a particular size, shape and other physical characteristics, it is limiting if one wishes to introduce additional functionality via the metal centres such as redox activity, magnetism, colour or luminescence: examples of cages where these characteristics are important are surprisingly limited.5We have recently been interested to include metal ions such as Ru(II) and Os(II) into coordination cage assemblies to exploit their well known redox and luminescence properties in coordination cages that consequently have a wider range of useful properties than simply the ability to bind guests.6 The reversible redox behaviour of these at modest potentials,6a,b and the availability of stable, long-lived MLCT excited states of an array of chromophores around the central cavity,6b make these particularly appealing metal ions which could allow (for example) a reversible change in the charge of a host cage, or the ability to effect photoinduced energy/electron transfer to a bound guest. However, these desirable properties are also associated with the high kinetic inertness of second- and third-row transition metal ions in a low-spin configuration, which makes Ru(II) and Os(II) very difficult to use in conventional self-assembly processes which rely on kinetic lability.
The consequence of this is that a more sophisticated synthetic strategy must be used to permit inclusion of kinetically inert metal ions in elaborate self-assembled polynuclear metal assemblies. The strategy is a stepwise ‘complexes as ligands’ approach that we6 and others7 have used. This involves initial preparation of a mononuclear complex of the kinetically inert metal ion but which bears pendant binding sites at which cage assembly can propagate. Combination of this ‘complex ligand’ with labile ions in a separate step results in completion of the cage assembly in which, necessarily, the labile and inert metal ions strictly alternate around the periphery. This is notably different from the use of unsymmetrical ligands, which possess both hard and soft binding sites which will selectively bind to hard and soft metals, respectively: this has been exploited by many groups to give mixed-metal cage assemblies but this method still requires both types of metal to be labile.8
Our recent efforts towards this end have focussed on the preparation of heterometallic [(Ma)4(Mb)4(Lnaph)12]X16 cubic coordination cages (where Ma = Os/Ru, and Mb = Co/Cd; see Scheme 1 for ligand structure); these were prepared from inert [(Ma)(Lnaph)3]2+ ‘complex ligands’ with three pendant binding sites arising from the ditopic ligands, by reaction with additional labile [Mb]2+ ions (Fig. 1).6a,b These structures are essentially the same as those of the homonuclear [M8(Lnaph)12]X16 parent cages, in which eight octahedral metals define the vertices of an approximate cube, and twelve bis-bidentate bridging ligands define the edges.9 Both Ru(II) and Os(II) impart redox activity to the cages, allowing the charge on the cage cation to be switched reversibly between 16+ and 20+. In addition the Os(II) tris(pyrazolyl-pyridine) units have a long-lived excited state which is good electron-donor, potentially allowing photoinduced electron transfer from the cage superstructure to bound guests.6b
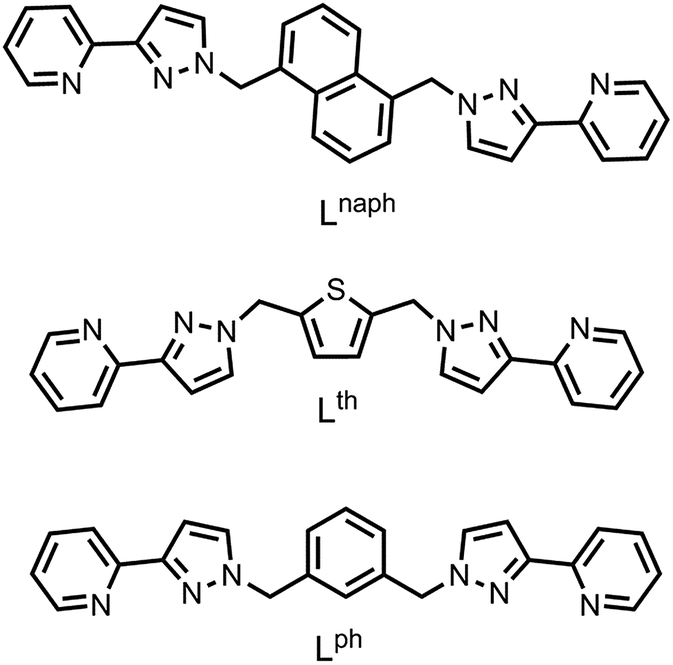 | ||
| Scheme 1 Structures of the ligands discussed in this work. | ||
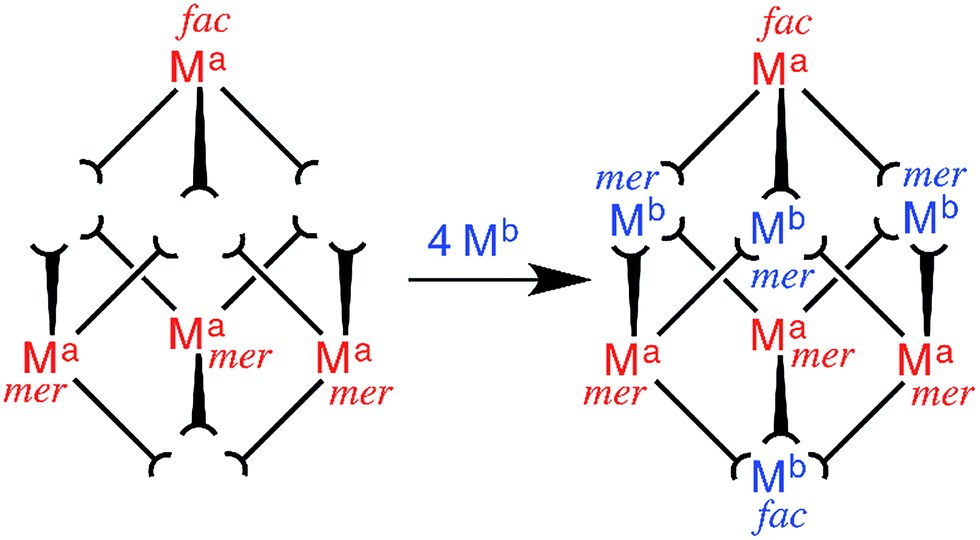 | ||
| Fig. 1 Sketch outlining the stepwise synthetic strategy used to prepare the heterometallic cubic cage complexes: viz combination of pre-formed, kinetically inert [(Ma)(Lnaph)3]2+ (Ma = Ru, Os) units with additional labile ions (Mb)2+ (Mb = Co, Cd) in a 4:4 ratio to give octanuclear [(Ma)4(Mb)4(Lnaph)12]16+.6a,b | ||
A subtle but crucial structural feature which allowed the stepwise assembles of these heterometallic cages to work is the geometric isomerism of the metal vertices.6b,7i These [M8(Lnaph)12]X16 cages possess two facial (fac) tris-chelate metal sites at opposite corners of a long diagonal of the cube. The six remaining metals all possess a meridional (mer) tris-chelate coordination geometry, such that the cages have overall molecular S6 symmetry. This particular combination of fac and mer metal centres arises spontaneously in the self assembly of these particular cages when labile metal ions such as Co(II) are used9 (in other types of cage this ratio may be different according to the requirements of each cage structure).1c Fortuitously, this 1:3 ratio of fac:mer metal complex units is also exactly what arises for simple statistical reasons when the mononuclear [(Ma)(Lnaph)3]2+ ‘complex ligands’ are prepared using Ru(II) or Os(II). This means that we can prepare mononuclear [(Ma)(Lnaph)3]2+ (Ma = Ru, Os) and use the 1:3 fac:mer mixture of geometric isomers directly, without separation, to complete the assembly of the heterometallic [(Ma)4(Mb)4(Lnaph)12]X16 cages which, precisely, require one of the four Ma sites to be fac and the other three to be mer.6a,b
In this contribution, we look at heterometallic assemblies containing Ru(II) ions as the inert component but based on different bridging ligands (Lph and Lth, with 1,3-benzene-diyl and thiophene-2,5-diyl spacers separating the two pyrazolyl-pyridine termini – see Scheme 1). These ligands have afforded some new heterometallic assemblies whose formation is controlled by the availability of different proportions of fac and mer mononuclear units, and include an unusual new type of heterometallic cubic cage which encapsulates both anions and cations in its central cavity.
Results and discussion
Synthesis and characterisation of [Ru(Lth)3](PF6)2
We have previously reported a series of molecular squares and coordination polymers with the thiophene-containing ligand Lth, in which the sulfur atom plays no part in the coordination chemistry but the thienyl unit just acts as a central spacer.10 For example in [M4(Lth)6]X8 (M = Co, Ni, Cu) there is a square array of M(II) ions, with the four edges of the square bridged alternately by one or two ligands Lth (Fig. 2a). In these [M4(Lth)6]X8 assemblies all metal centres have the mer tris-chelate coordination geometry, as this is what the self-assembly process using labile M(II) ions selects.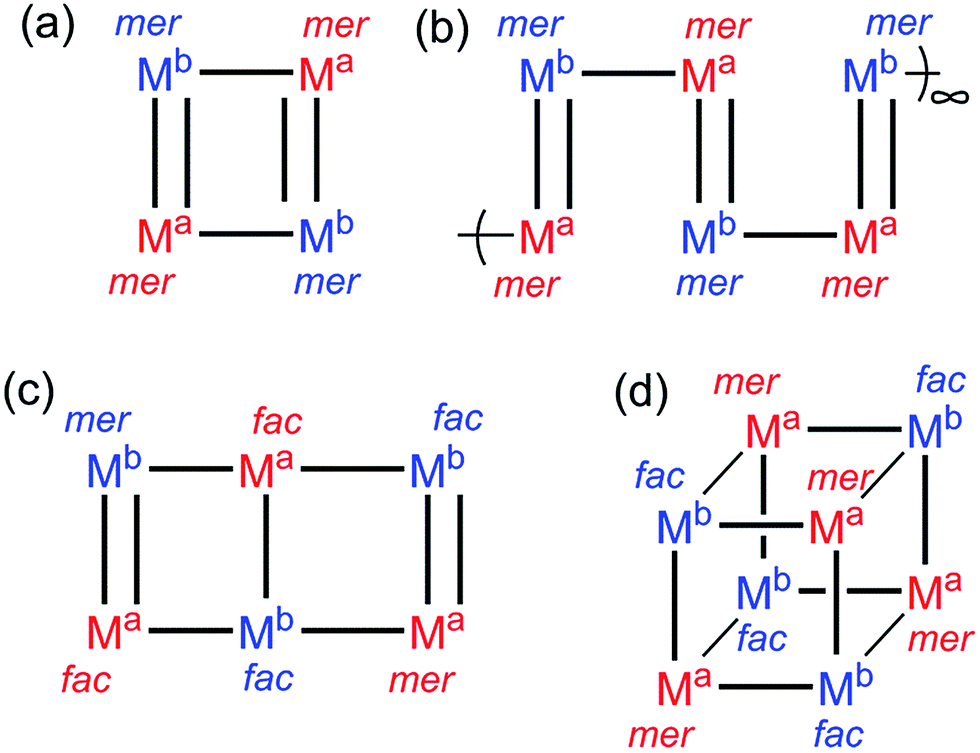 | ||
| Fig. 2 Summary of the types of assembly discussed in this paper: black lines denote bridging ligands; the chemically different types of metal ion (Ma and Mb) are colour coded; and the fac or mer geometry at each position is indicated. | ||
Therefore, the question is: if an inert, pre-formed Ru(II) complex containing a mixture of fac and mer isomers is used in the assembly, would it afford a different product due to the constraint that some fac complex units must be present; or will the mer Ru(II) units be selected, and the fac units simply be ignored and excluded from the self-assembly process?
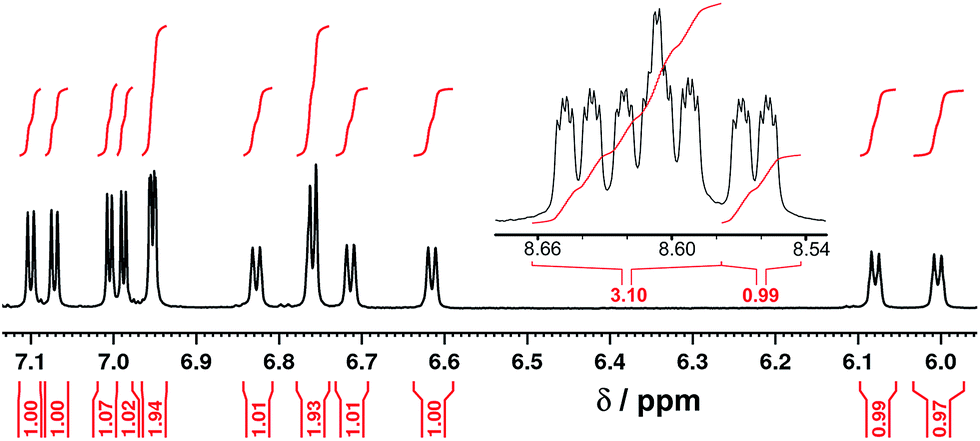
The mononuclear complex ligand [Ru(Lth)3](PF6)2 was prepared by reaction of >3 equivalents of Lth with one equivalent of Ru(dmso)4Cl2 in ethylene glycol at reflux, followed by anion metathesis and chromatographic purification during which the product was isolated as a single fraction with no apparent separation of fac and mer isomers. The ES mass spectrum confirmed the formation of the desired complex. The 1H NMR spectrum of [Ru(Lth)3](PF6)2 showed that the expected11 1:3 fac:mer ratio of geometric isomers has formed. In the threefold-symmetric fac isomer all three ligands are equivalent, but this product is only one third as abundant as the mer isomer in which all three ligands are inequivalent. The result is the presence of four ligand environments with equal abundance, which the 1H NMR spectrum shows clearly (Fig. 3 and 4).
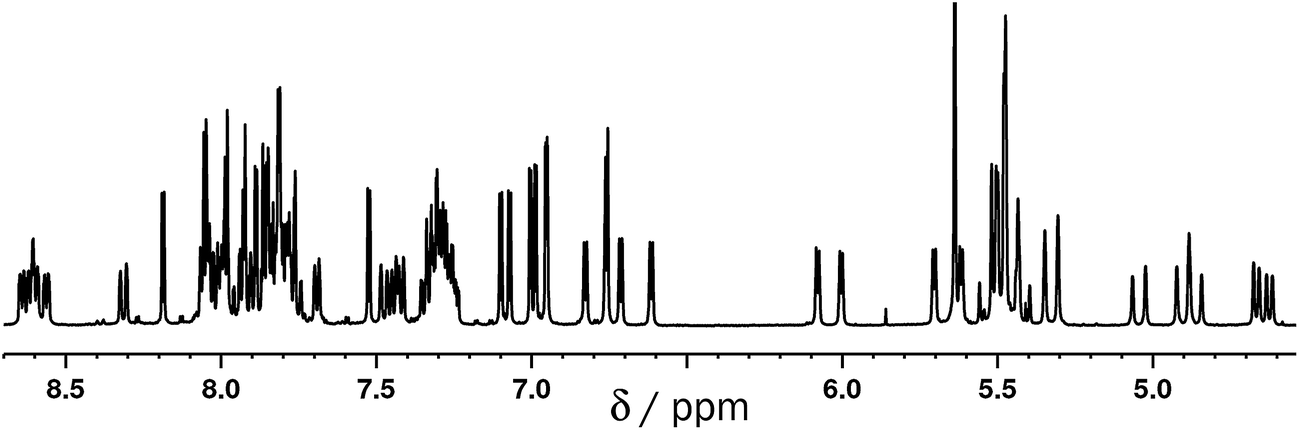 | ||
| Fig. 3 1H NMR spectrum (400 MHz, CD3CN) of [Ru(Lth)3](PF6)2. | ||
 | ||
| Fig. 4 Expansions of the 1H NMR spectrum (400 MHz, CD3CN) of [Ru(Lth)3](PF6)2 in Fig. 3. The 3:1 mer/fac mixture of isomers is clear from the presence of four different environments for each proton type with equal intensity, e.g. the expansion of the region around 8.6 ppm for coordinated pyridyl H6 protons showing the presence of four doublets (with additional fine structure). | ||
Mixed metal structures incorporating [Ru(Lth)3](PF6)2 units
This mixture of geometric isomers for [Ru(Lth)3](PF6)2 does not provide exactly what is required for assembly of the complete squares (Fig. 2a) and chains (Fig. 2b) obtained using labile first-row metal ions, in which only the mer isomer is used.9 Reaction of [Ru(Lth)3](PF6)2 (3:1 mixture of mer:fac isomers) with excess Co(BF4)2 (4.7 eq.) in methanol/dichloromethane solution instantly precipitated a yellow powder which was collected and thoroughly washed with methanol and dichloromethane, before recrystallisation from acetonitrile/ether to yield a crystalline product as fine yellow needles. Whereas the ES mass spectrum of the crude reaction mixture indicated the presence of RuL3, RuCoL3 and Ru2Co2L6 species in solution, the X-ray crystal structure identified the structure of the product as the molecular square [Ru2Co2(Lth)6](BF4)5(PF6)3·2.5MeCN·H2O (Fig. 5 and 6) which has been able to select the best combination of anions from the mixture present to facilitate crystallisation.
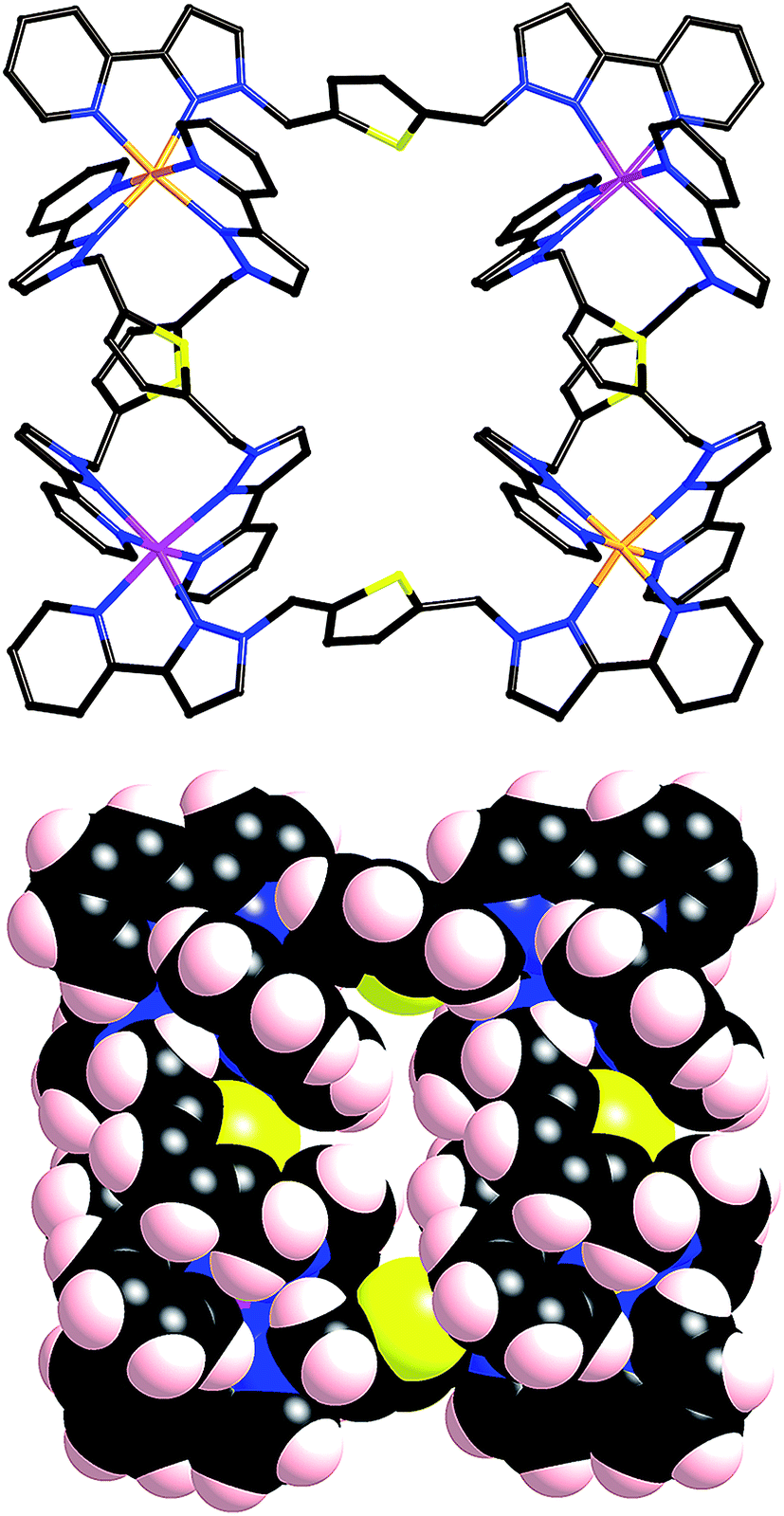 | ||
| Fig. 5 Two views of the complete complex cation from the X-ray crystal structure of [Ru2Co2(Lth)6](BF4)5(PF6)3. Metal atom positions have been coloured differently to emphasise the alternating arrangement within each complex molecule, but disorder means that each metal atom site refines best as a 50:50 mixture of Ru and Co. | ||
 | ||
| Fig. 6 Two views emphasising the interaction of one BF4− and one PF6− anions with the complex cation in the X-ray crystal structure of [Ru2Co2(Lth)6](BF4)5(PF6)3. | ||
The structure is essentially the same as those of the homonuclear squares reported previously.10 It consists of two homochiral M2(Lth)2 double helical units which are crosslinked by additional ligands to give the approximately square structure (with an alternating sequence of two and one bridging ligands spanning the edges, Fig. 2a). M⋯M separations are in the range 8.9–11.1 Å and M–M–M angles at the corners of the ‘square’ lie in the range 89.1–90.9°. All four metal centres have a mer tris-chelate coordination geometry. Due to the stepwise nature of the synthesis, in which every pendant pyrazolyl-pyridine binding site from the [Ru(Lth)3]2+ units must bind to a Co(II) ion, we must have an alternating sequence of Ru(II) and Co(II) ions around the periphery of the square. This could adopt two possible orientations in the crystal: if the metal sites are labelled sequentially 1-2-3-4 around the ring we could have Ru(1)/Co(2)/Ru(3)/Co(4) or Co(1)/Ru(2)/Co(3)/Ru(4), with the difference in the scattering power of Ru and Co atoms making them easily distinguishable by X-ray crystallography. However it appears that the structure is crystallographically disordered with the arrangements superimposed such that every metal atom site is best refined as 50% Ru and 50% Co. This is presumably facilitated by the similar coordination environments around the Ru(II) and Co(II) ions such that the ligand atoms appear in the same position if the metal ions are swapped over: thus only the metals are disordered, the ligand atom positions are not significantly affected by swapping the metal atom positions. This has been observed in other Ru(II)/Co(II) systems we have reported previously.6b
Two anions (PF6− and BF4−) sits on either side of the central region of the square, where there is a ‘nest’ of inwardly directed protons, forming numerous C–H⋯F hydrogen-bonding interactions (Fig. 6). The sulphur atoms of the thiophene rings apparently do not form any intermolecular interactions; there are instead, as with the homonuclear squares, intramolecular interactions between the exocyclic lone pairs and (electron-deficient) coordinated pyrazolyl rings on adjacent ligands in the helical M2L2 units.
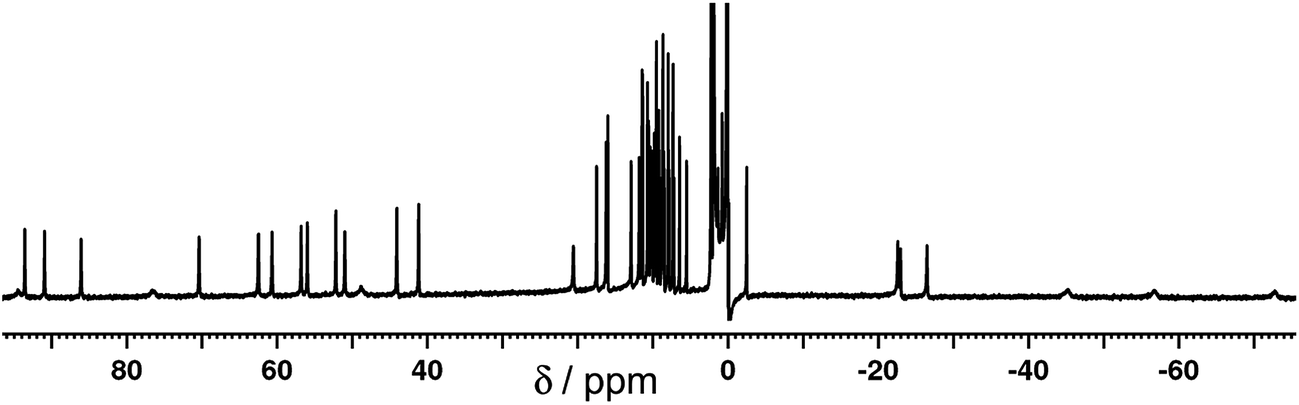
The 1H NMR spectrum of the redissolved crystals indicates that the structure observed in the solid state is preserved in solution (Fig. 7 and 8). Due to the paramagnetism of the high-spin Co(II) centres, the signals are shifted over the range of +100 to −80 ppm, as we have seen numerous times with structures of this type.9 In homonuclear [Co4(Lth)6](BF4)4, 27 1H NMR signals were observed in the NMR spectra, indicating 1.5 inequivalent ligand environments in agreement with the crystallographic symmetry.10 However, with alternating Ru(II) and Co(II) centres in the mixed-metal complex Ru2Co2 complex we have lost a twofold symmetry element, resulting in three inequivalent ligand environments, each with no internal symmetry, and therefore we expect 54 independent proton resonances. Of these we expect those close to Co(II) to be most affected by the paramagnetism (broadened and/or shifted), and the protons close to the Ru(II) centres to be less affected.
 | ||
| Fig. 7 1H NMR spectrum (400 MHz, CD3CN) of [Ru2Co2(Lth)6](BF4)4(PF6)4. | ||
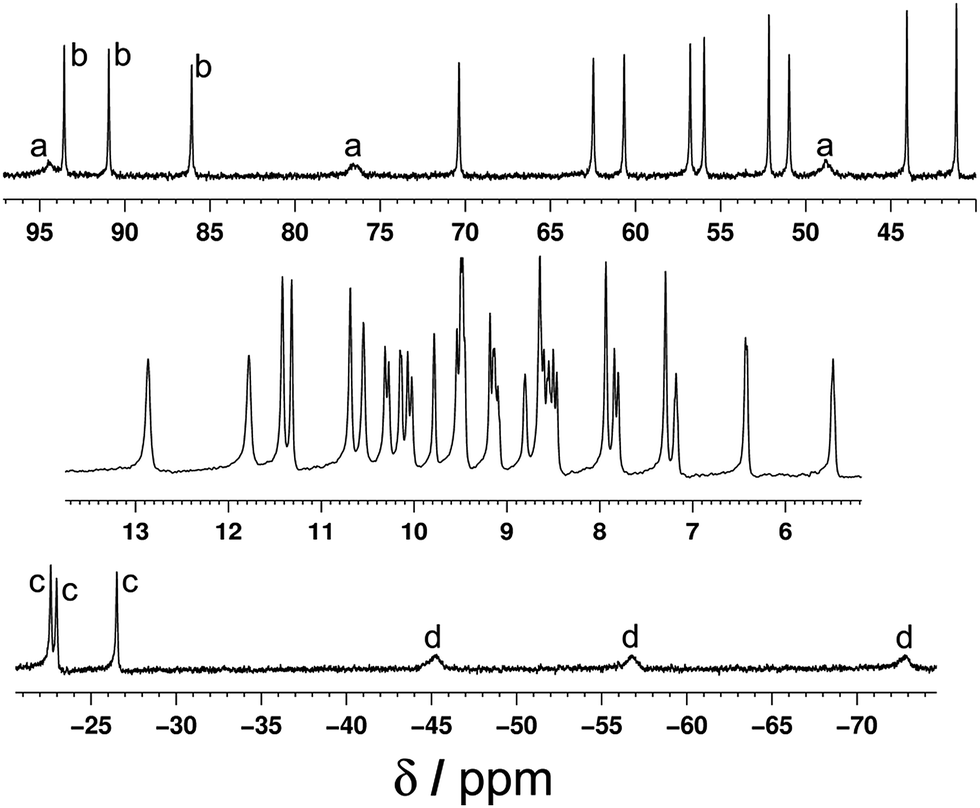 | ||
| Fig. 8 Expansions of the 1H NMR spectrum (400 MHz, CD3CN) of [Ru2Co2(Lth)6](BF4)4(PF6)4 (most but not all regions of the spectrum in Fig. 7 are included). The labels a–d denote sets of three ligands corresponding to a particular ligand proton in each of three different environments. | ||
This is apparent in the expansions in Fig. 8 in which we can see exactly the expected number of signals, split into two groups. Half of the signals occur in the 0–12 ppm region, from protons which are close to the Ru(II) but remote from Co(II); in some cases the fine coupling that is normal in spectra of diamagnetic compounds but usually lost for paramagnetic compounds is retained. The other half of the signals are far more widely dispersed (>15 and <−20 ppm) and arise from the protons closer to Co(II). In addition we can see in several places that the signals clearly come in sets of three, corresponding to the three ligand environments (e.g. the three broad signals between −40 and −80 ppm, and the three sharp signals between −20 and −30 ppm). Some of these are labelled in Fig. 8. Overall this spectrum clearly confirms that the structure observed in the solid state is retained in solution.
The DOSY spectrum in the 0–12 ppm region was measured, giving a single diffusion constant for all observed protons [logD (m2 s−1) = −9.2] that is characteristic of a large polynuclear assembly9 and clearly not characteristic of a mononuclear complex.6c The mass spectrum of redissolved crystals showed that some fragmentation occurred under the mass spectral conditions; a series of peaks corresponding to {RuCo(Lth)3}n+ species was observed, but importantly a series of peaks for the intact cation {Ru2Co2(Lth)6X8−n}n+ (with loss of varying numbers of anions) was also present.
Reaction of [Ru(Lth)3](PF6)2 (3:1 mixture of mer:fac isomers) with excess Cd(ClO4)2 (5.7 eq.) in methanol/dichloromethane solution instantly precipitated a yellow powder which was collected and thoroughly washed with methanol and dichloromethane, before recrystallisation from acetonitrile/ether to yield the product as fine yellow needles which gave analytical data consistent with the formulation [CdRu(Lth)3](ClO4)2(PF6)2. The ES mass spectrum is consistent with this, showing main signals corresponding to the [CdRu(Lth)3]4+ cation associated with varying numbers of anions; the isotope pattern further confirms the formulation.
We expect this species this to have a similar structure to the homometallic Cd(II) complex {[Cd2(Lth)3]X4}∞, which is a one-dimensional coordination polymer consisting of an infinite chain of Cd(II) ions with an alternating arrangement of two and one bridging ligand between each adjacent pair of Cd(II) ions, as sketched in Fig. 2b: effectively, a linear chain of double helical {Cd2(Lth)2}4+ units connected end-to-end by additional Lth units which complete the sixfold coordination around each Cd(II) ion.10 The mer tris-chelate geometry around every Cd(II) ion means that all three ligands are inequivalent. Consistent with this, the 1H NMR spectrum of redissolved crystals of [CdRu(Lth)3](ClO4)2(PF6)2 revealed the presence of three independent ligand environments, each with no internal symmetry (Fig. 9) due to the inequivalence of Ru(II) and Cd(II) at either end of each ligand. For example it is apparent from the COSY spectrum that there are three pairs of doublets from the thienyl rings and six pairs of doublets from diastereotopic CH2 groups (Fig. 9). Unfortunately, the crystals were extremely thin and weakly diffracting and the resultant structure is not of publishable quality, but it was sufficient to confirm that our assumption about the structure is correct: it is indeed a one-dimensional coordination polymer {[CdRu(Lth)3](ClO4)2(PF6)2}∞, similar to the homometallic Cd(II) analogue10 but with (necessarily) an alternation of Ru(II) and Cd(II) ions along the chain.
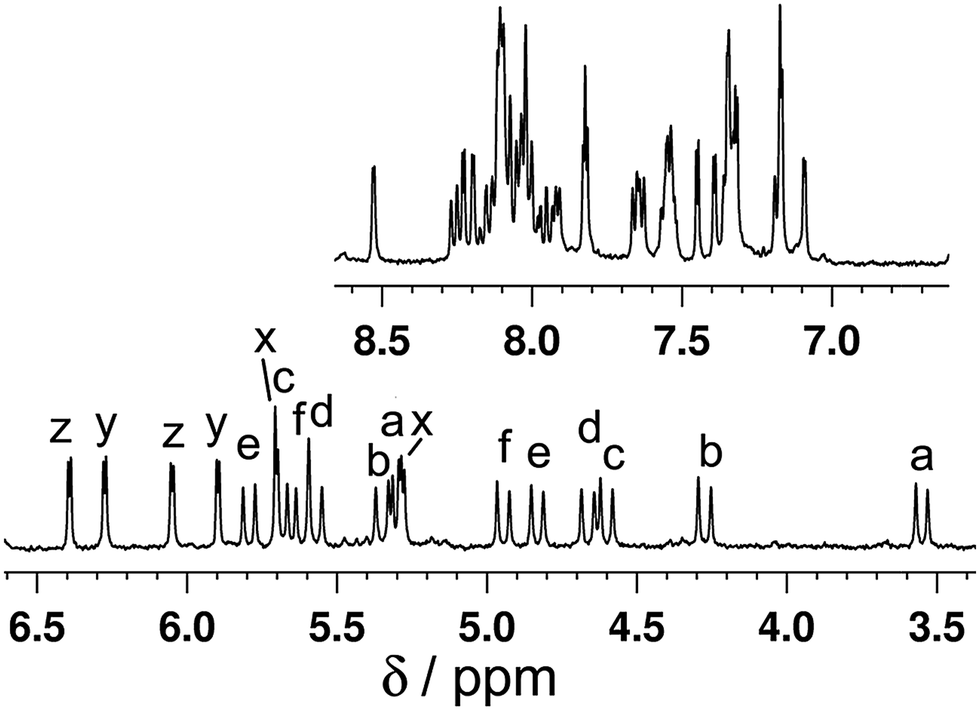 | ||
| Fig. 9 1H NMR spectrum (400 MHz, CD3CN) of {[CdRu(Lth)3](ClO4)2(PF6)2}∞. The six pairs of doublets from diastereotopic CH2 groups are labelled a–f; the three pairs of doublets from the thienyl rings (with much smaller coupling constants) are labelled x, y and z. These assignments were made from a COSY spectrum and confirm that the complex in solution has three independent ligand environments, each with no internal symmetry, as required for the structural type in Fig. 2b. | ||
Overall, even though a 3:1 mer:fac mixture of isomers of the relevant [Ru(Lth)3]2+ building block was used, in both new examples shown here only the mer isomer was selected for incorporation into the mixed-metal assemblies – the fac [RuL3]2+ units are not used.
Synthesis and characterisation of [Ru(Lph)3](PF6)2
The ligand Lph has also been studied before: reaction of Lph with transition metal dications in a 3:2 ratio leads to formation of approximately cubic [M8(Lph)12]X16 cages which have the same type of S6-symmetric metal framework as seen in the cages [M8(Lnaph)12]X16: viz. two metal ions at either end of a long diagonal of the cube have a fac tris-chelate coordination environment whereas the other six have a mer geometry (i.e. a 3:1 mer/fac ratio), with an inversion centre meaning that the cage as a whole is achiral.9 In some cases we also observed formation of lower-symmetry [M6(Lph)9]X12 assemblies which have a core structure reminiscent of a slightly bent ‘open book’ with metal ions at the four vertices and either end of the central spine, with bridging ligands arrayed along the edges [Fig. 2, structure (c)]. In these cases four of the six metal vertices (the central two and two at diagonally opposed corners) have a fac tris-chelate structure, with the other two metal vertices (the remaining two corners) having a mer tris-chelate geometry, giving a mer:fac ratio of 1:2. We might expect, therefore, that [Ru(Lph)3]2+ units could be incorporated into either or both of these types of assembly: either M8L12 (Fig. 1) or M6L9 (Fig. 2c) depending on the relative amounts of mer and fac [Ru(Lph)3]2+ units that are available from its synthesis.
[Ru(Lph)3](PF6)2 was prepared by reaction of RuCl2(dmso)4 with >3 equiv. Lph in refluxing ethylene glycol, and after work-up a yellow solid was isolated whose analytical and ES mass spectrometric data were consistent with the formulation [Ru(Lph)3](PF6)2. Interestingly, 1H NMR spectroscopic analysis showed that the mixture was not formed as the expected statistical 3:1 mer/fac mixture: instead, the mixture contained an approximately 4:1 mer/fac ratio (Fig. 10 and 11). In areas where the separate peaks are clearly resolved we can identify three closely-spaced signals with an arbitrary intensity of 1.0 (corresponding to the three different ligand environments of the mer isomer), and a fourth signal (from the fac isomer) which has a relative intensity of approximately 0.72. This gives a mer/fac ratio of approximately 4.2:1. In this case we suggest that steric interactions between the three ligands, which will be more severe in the fac isomer, are sufficiently significant to give an excess of the kinetically favoured mer isomer compared to what is statistically expected (Fig. 11).11
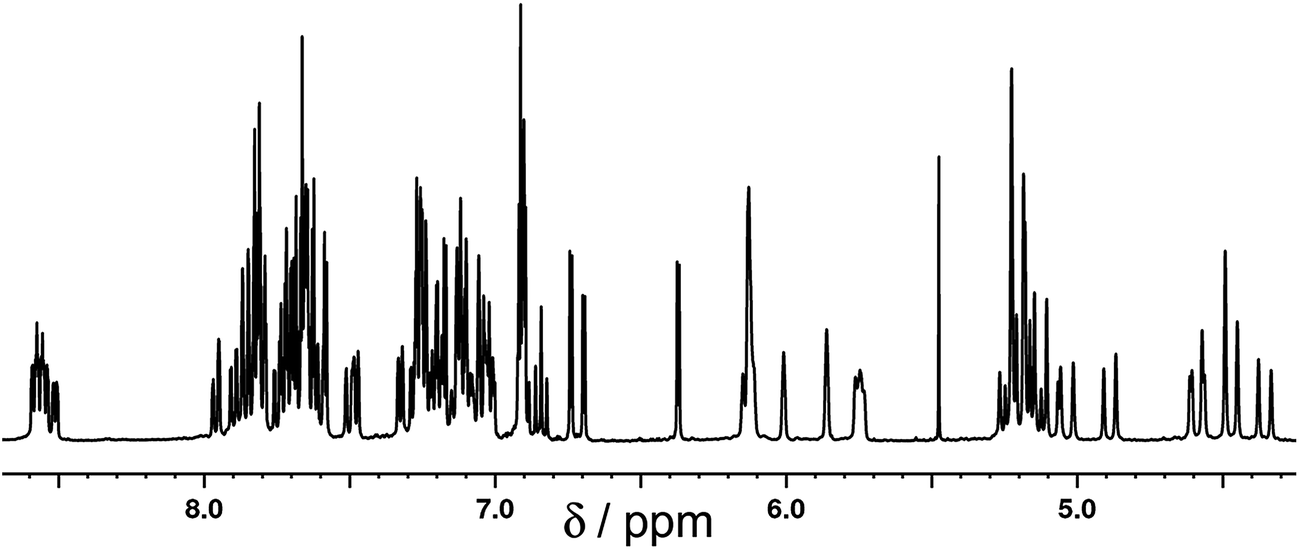 | ||
| Fig. 10
1H NMR (CD3CN, 400 MHz) spectrum of [Ru(Lph)3](PF6)2 as a 4:1 mixture of mer:fac isomers (the peak at 5.5 ppm is a trace of CH2Cl2). | ||
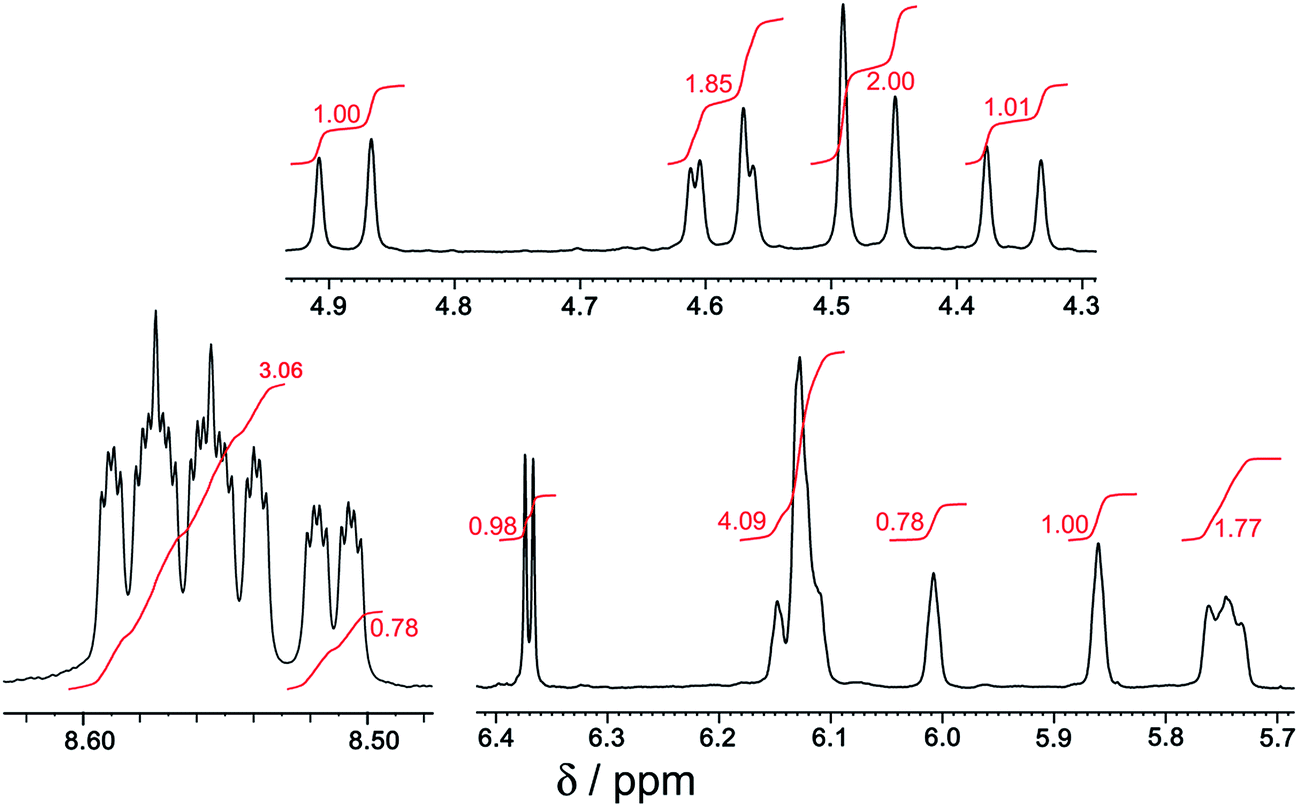 | ||
| Fig. 11 Expansion of parts of Fig. 10; numbers in red are integral values. | ||
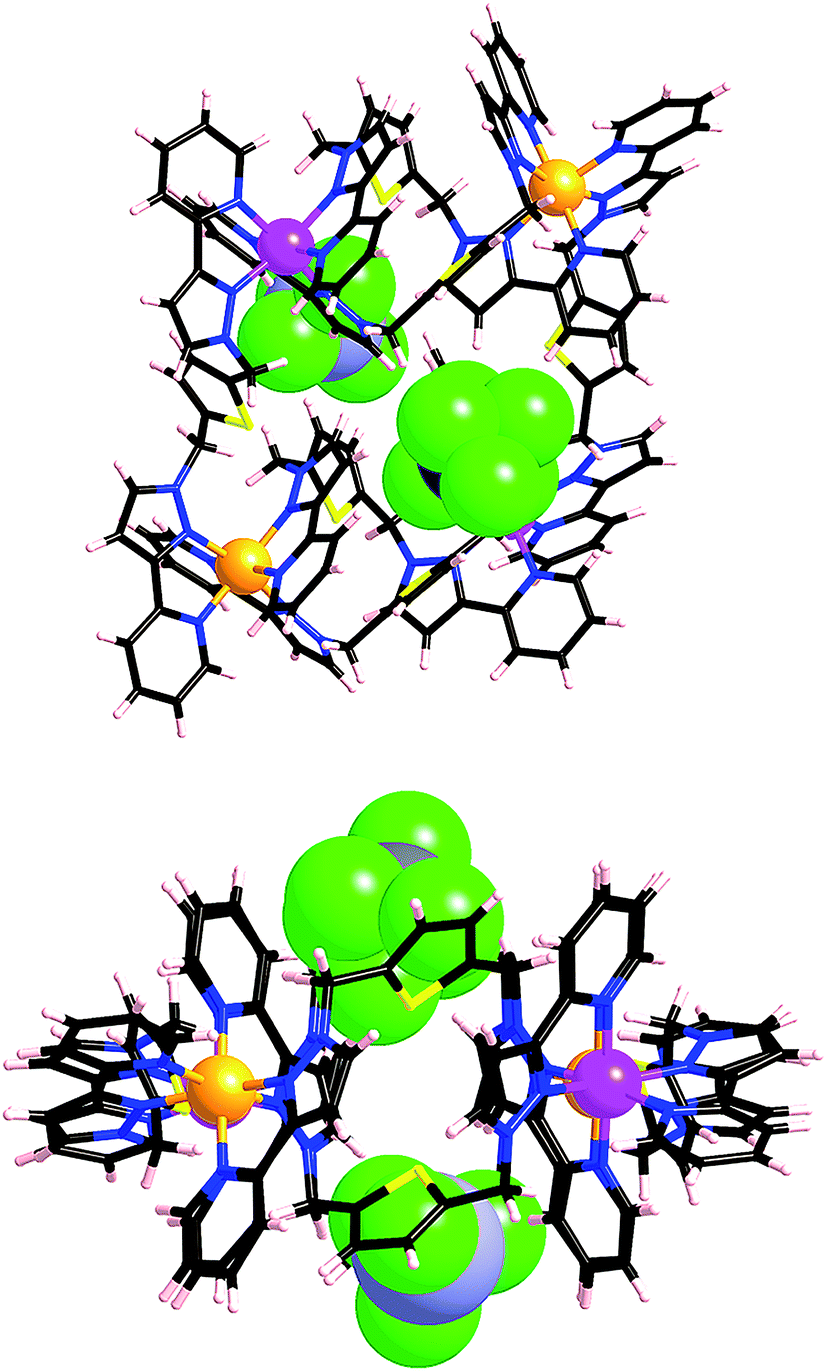
This 4:1 mer:fac ratio of vertices has not been observed in any of the structures we have reported to date. We were therefore interested to see what types of heteronuclear assembly could be prepared using our as-isolated [Ru(Lph)3](PF6)2 sample. Accordingly, [Ru(Lph)3](PF6)2 (ca. 4:1 mixture of mer:fac isomers) was combined with Co(BF4)2 in dichloromethane/methanol solution. After filtration and washing, the resultant precipitate was recrystallized from acetonitrile, with slow diffusion of diisopropyl ether vapour into the solution yielding yellow X-ray quality crystals. The structural determination revealed the structure to be a [Ru3Co3(Lph)9](BF4)12 ‘open book’ assembly (Fig. 12), which is structurally analogous to the homonuclear [M6(Lph)9]12+ assemblies that we have seen before.12
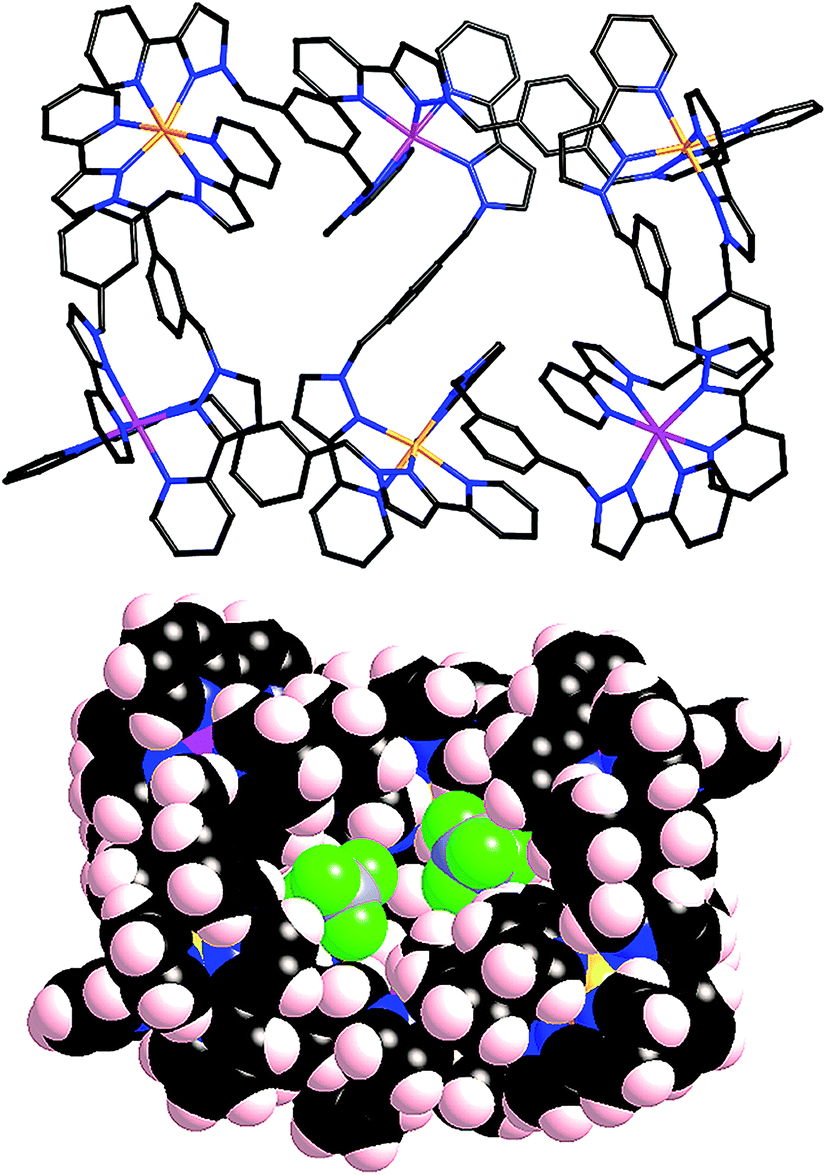 | ||
| Fig. 12 Two views of the structure of [Ru3Co3(Lph)9](BF4)12. Left: a view of the complete complex cation (all metal sites are 50:50 disordered, but in each molecule the metal ions necessarily alternate as indicated by the pink/orange colours). Right: a space-filling view emphasising the interaction of two anions with the complex cation. | ||
The six metal ions are arranged in the manner of two squares sharing one edge, drawing comparison to an ‘open book’ structure. Both pairs of metal atoms forming outer edges of the ‘book’ are connected by two ligands in a double helical strand; four more ligands connect the outer metals to the ‘spine’ of the book, with the final ligand forming the ‘spine’ itself. Ru(II) and Co(II) ions necessarily occupy alternating sites within the framework, which again leads to two possible orientations of the heterometallic structure in the crystal. Again these are disordered such that unambiguous crystallographic labelling of each metal-ion site is not possible, but each site is refined as 50:50 Ru:Co. This is reflected in a moderate shortening of the metal–nitrogen bond lengths compared to what we observed in [Co6(Lph)9](BF4)12: an average M–N bond length of 2.09 Å is observed (ranging between 2.05–2.13 Å), compared to 2.12 Å in [Co6(Lph)9](BF4)12. The M⋯M separations around the edge of the ‘book’ are in the range 9.58–9.73 Å, and along the ‘spine’ the separation is 10.50 Å. The angle between the two ‘pages’ of the book (i.e. between the two M4 squares) is ca. 125°, resulting in two bowl-like cavities in which sit BF4− anions stabilised by numerous CH⋯F interactions (Fig. 12 and 13).
 | ||
| Fig. 13 Two additional views of the structure of [Ru3Co3(Lph)9](BF4)12 emphasising the CH⋯F interactions between the two closely-associated BF4− anions and the complex cation. | ||
1H NMR spectroscopy was of limited use due to the low symmetry. Homonuclear complexes [M6(Lph)9]X12 possess only a C2 axis in solution such that there are 4.5 independent ligand environments leading to 89 signals of relative intensity 2H and two (on the C2 axis) of intensity 1H.12b In the mixed-metal complex this twofold symmetry is lost, such that we expect 180 independent 1H signals, affording a highly complex NMR spectrum that cannot be meaningfully interpreted. However, ES mass spectrometry again confirmed the structural integrity of the complex in solution with signals corresponding to {[Ru3Co3(Lph)9](BF4)12−n}n+, i.e. the complete complex cation associated with varying numbers of anions, being observed with the correct m/z value and isotopic patterns.
In this structure four of the six metal sites have a fac tris-chelate geometry, i.e. of the three [Ru(Lph)3]2+ units that are incorporated, two are fac and one is mer, despite the excess of the mer isomer of [Ru(Lph)3]2+ in the sample used to generate the assembly. It follows that formation of [Ru3Co3(Lph)9](BF4)12 does not make full use the available Ru(II) building blocks (as evidenced by the low isolated yield of the crystalline product): this contrasts with formation of the cubic cages [Ru4M4(Lnaph)12]X16 where the supply of fac and mer [Ru(Lnaph)3]2+ units is exactly in the 1:3 proportion required for the cage assembly to complete.9 For this reaction of [Ru(Lph)3](PF6)2 with Co(II) ions we assume that the remaining mer isomer of [Ru(Lph)3](PF6)2 that is not required to assemble the ‘book’ structure forms some other heterometallic assembly with Co(II) ions but we were unable to establish its identity: ES mass spectra of the remaining solution after separation of crystalline [Ru3Co3(Lph)9](BF4)12 showed only mononuclear complex species with no clear evidence for a larger assembly.
We were interested to see if we could isolate different assemblies containing [Ru(Lph)3]2+ units by changing the mer:fac ratio. Fletcher and co-workers demonstrated that the mer:fac ratio of a [RuL3]2+ complex based on a non-symmetrical chelating ligand can be skewed in favour of the mer isomer by performing the complexation under milder reaction conditions.11 So we repeated the synthesis of [Ru(Lph)3](PF6)2 at a much lower temperature, using refluxing ethanol/water mixture instead of refluxing ethylene glycol. After work-up a yellow solid was isolated which again analysed as [Ru(Lph)3](PF6)2 but this time 1H NMR analysis showed that it contained an approximately 7:1 mer/fac ratio of geometric isomers. Clearly, at lower temperature the reaction favours the kinetically more stable mer isomer. This does not make a huge difference to the isomeric composition which has changed from 80:20 mer:fac (preparation in ethylene glycol) to approximately 87:13 mer:fac (preparation in aqueous ethanol) but nonetheless this might affect the course of the assembly with Co(II) to give a heteronuclear species.
[Ru(Lph)3](PF6)2 (7:1 mixture of mer:fac isomers) was reacted with one equivalent of Co(BF4)2 in dichloromethane/methanol at room temperature overnight. After workup, a yellow solid was collected which was slowly recrystallized from nitromethane by vapour diffusion with THF. This mixture was monitored by ES mass spectrometry over the course of two months whilst the recrystallization was occurring, revealing an interesting product evolution. Initially the spectrum was dominated by signals for a dinuclear species {[CoRu(Lph)3]X2}2+ peaks [m/z 751, 785, 814; X = PF6, BF4 or F; Fig. 14a], but after a week, a series of peaks corresponding to the tetranuclear {[Co2Ru2(Lph)6]X5}3+ appeared [m/z 1036, 1055, 1075, 1094 for the different anions; Fig. 14b] which we assume to be a square like that in Fig. 5. Finally, after several months, a series of peaks corresponding to octanuclear {[Co4Ru4(Lph)12]X16−n}n+ had appeared [m/z 806, 942, 1123, 1377, 1757 for n = 8–4, respectively; Fig. 14c and 15]. Clearly assembly of the higher nuclearity species is slow under these conditions.
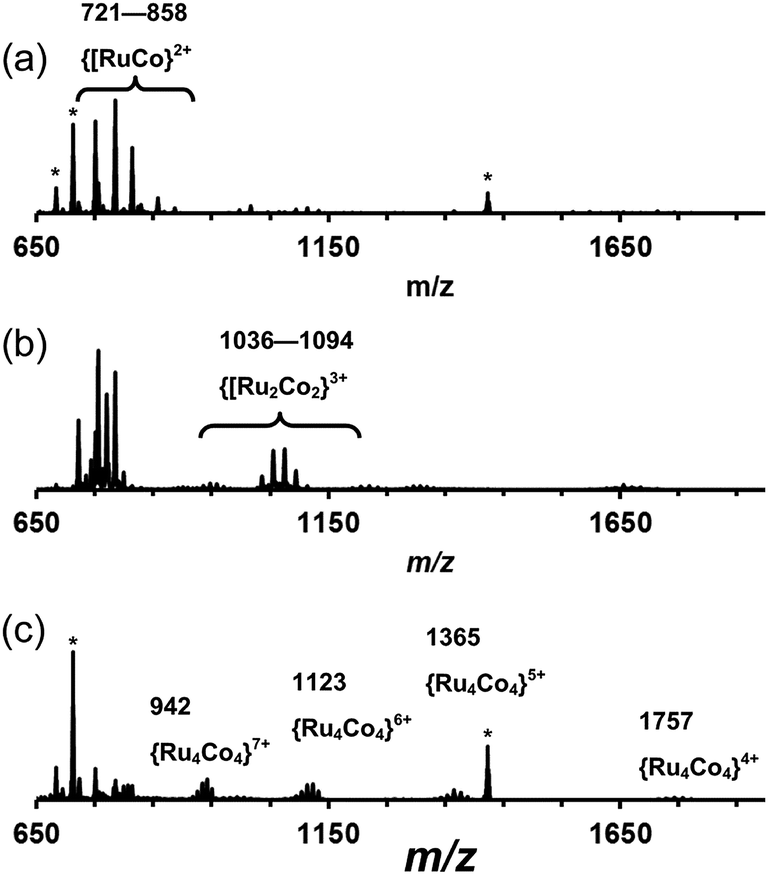 | ||
| Fig. 14 Series of ES mass spectra following the evolution of the product mixture of [ConRun(Lph)3n]X4n. (a) Product mixture after 1 day; (b) product mixture after 1 week; (c) product mixture after 2 months. Peaks labelled * are due to [Ru(Lph)3]2+, arising from fragmentation of the larger complexes. The presence of several closely-spaced peaks for each type of assembly arises because of the presence of a mixture of anions in solution. | ||
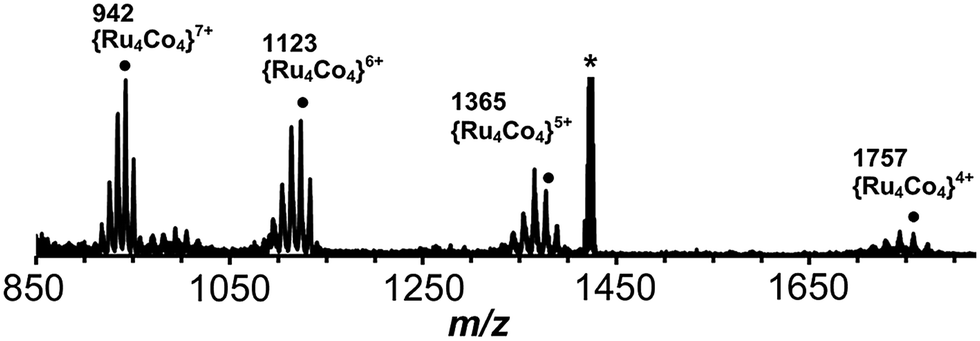 | ||
| Fig. 15 Expansion of the ES mass spectrum of [Co4Ru4(Lph)12]X16 (see Fig. 14c). The m/z values given are for the peaks labelled ● for which X16−n = {(PF6)15(BF4)} − nPF6. | ||
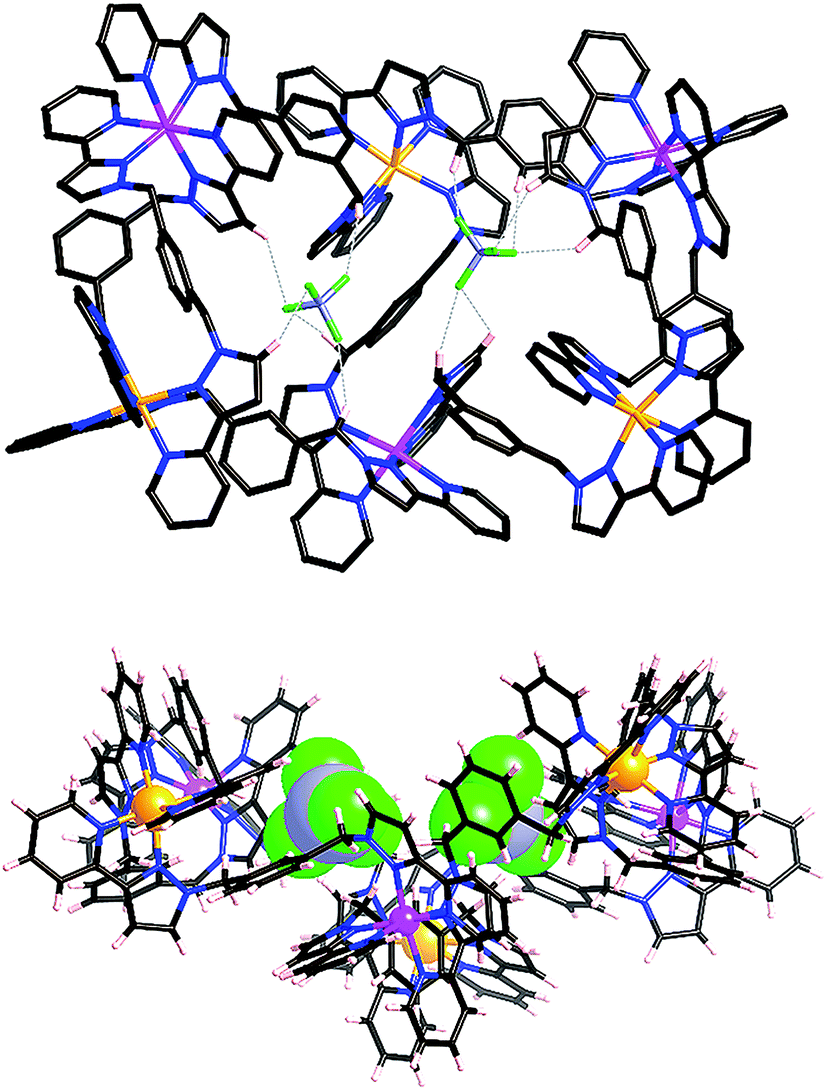
After several months, this solution yielded a crop of crystalline yellow blocks and orange shards. The yellow blocks were more abundant and of excellent X-ray quality. The structure revealed an octanuclear coordination cage cation, as expected on the basis of the mass spectrum, but with the formulation [Ru4Co4(Lph)12{Na(BF4)4}](PF6)6(BF4)7 (Fig. 16–18), i.e. containing an additional sodium cation and an associated anion. The Ru4Co4 metal framework is approximately cubic, with alternating Ru(II) and Co(II) ions at each metal site, as expected. Ru⋯Co separations along the edges are in the range 9.79–10.63 Å; M–M–M angles are in the range 80.0–103.0°. However the framework type is unexpectedly different from any type of cubic coordination cage that we have seen before.
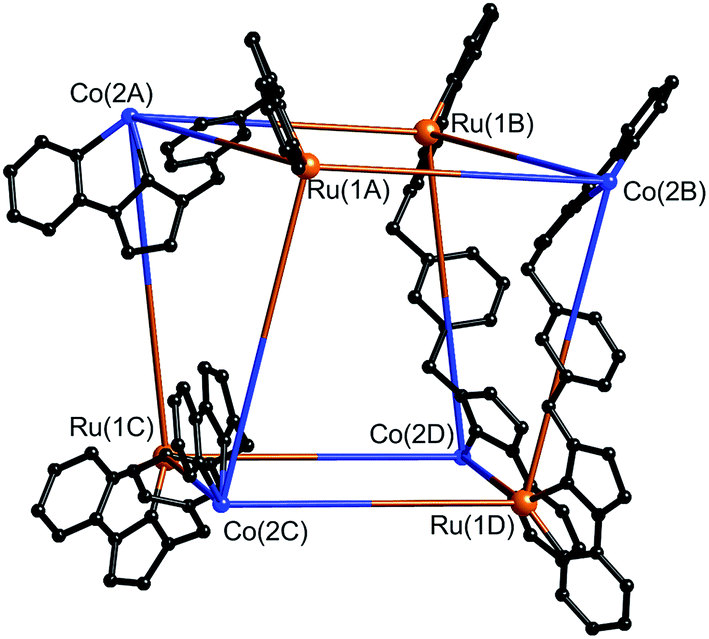 | ||
| Fig. 16 Partial view of the complex cation of [Ru4Co4(Lph)12{Na(BF4)4}](PF6)6(BF4)7. All Ru atoms and all Co atoms are crystallographically equivalent, but the two metal ion types are not disordered. Only four (crystallographically equivalent) ligands are shown. | ||
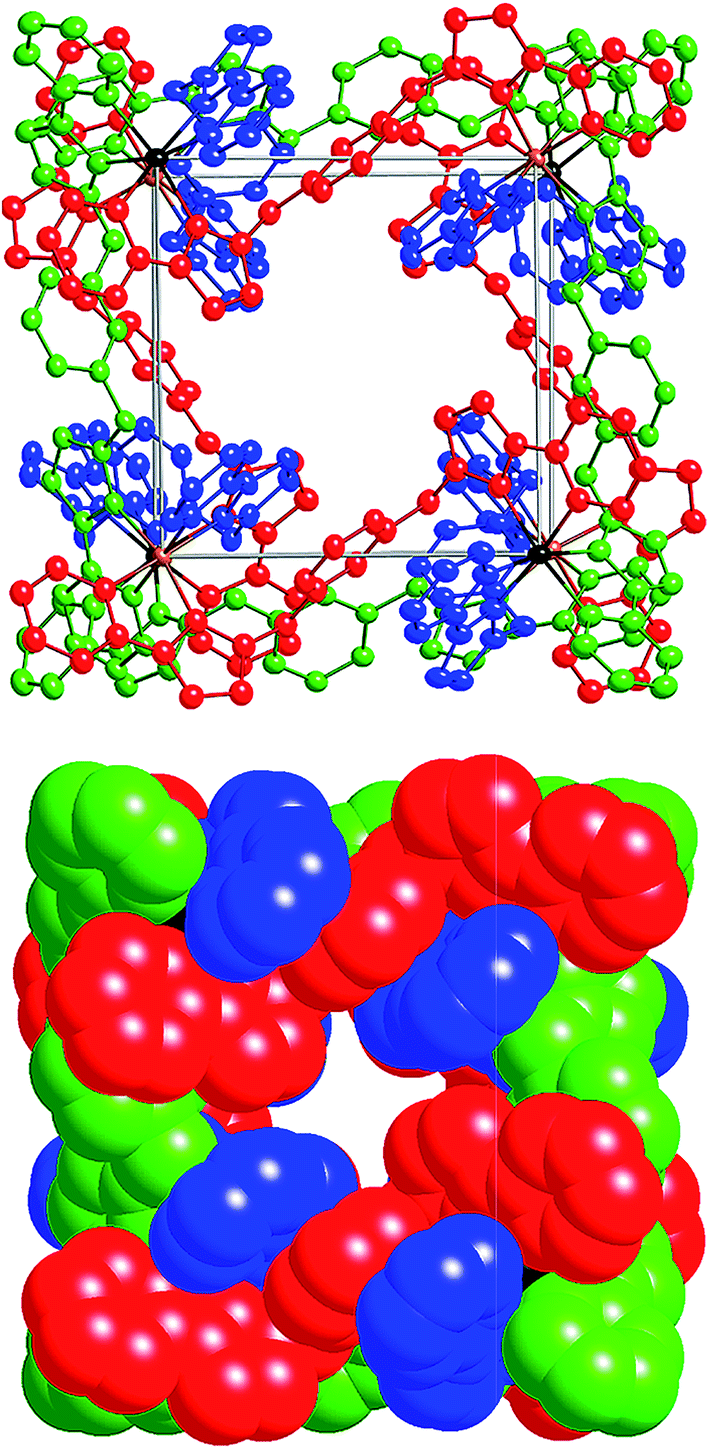 | ||
| Fig. 17 Left: View of the complete complex cation of [Ru4Co4(Lph)12{Na(BF4)4}](PF6)6(BF4)7. Thermal ellipsoids shown at 40% probability, and crystallographically equivalent ligands are coloured the same. Right: Space-filling view of the complex cation from the same perspective. | ||
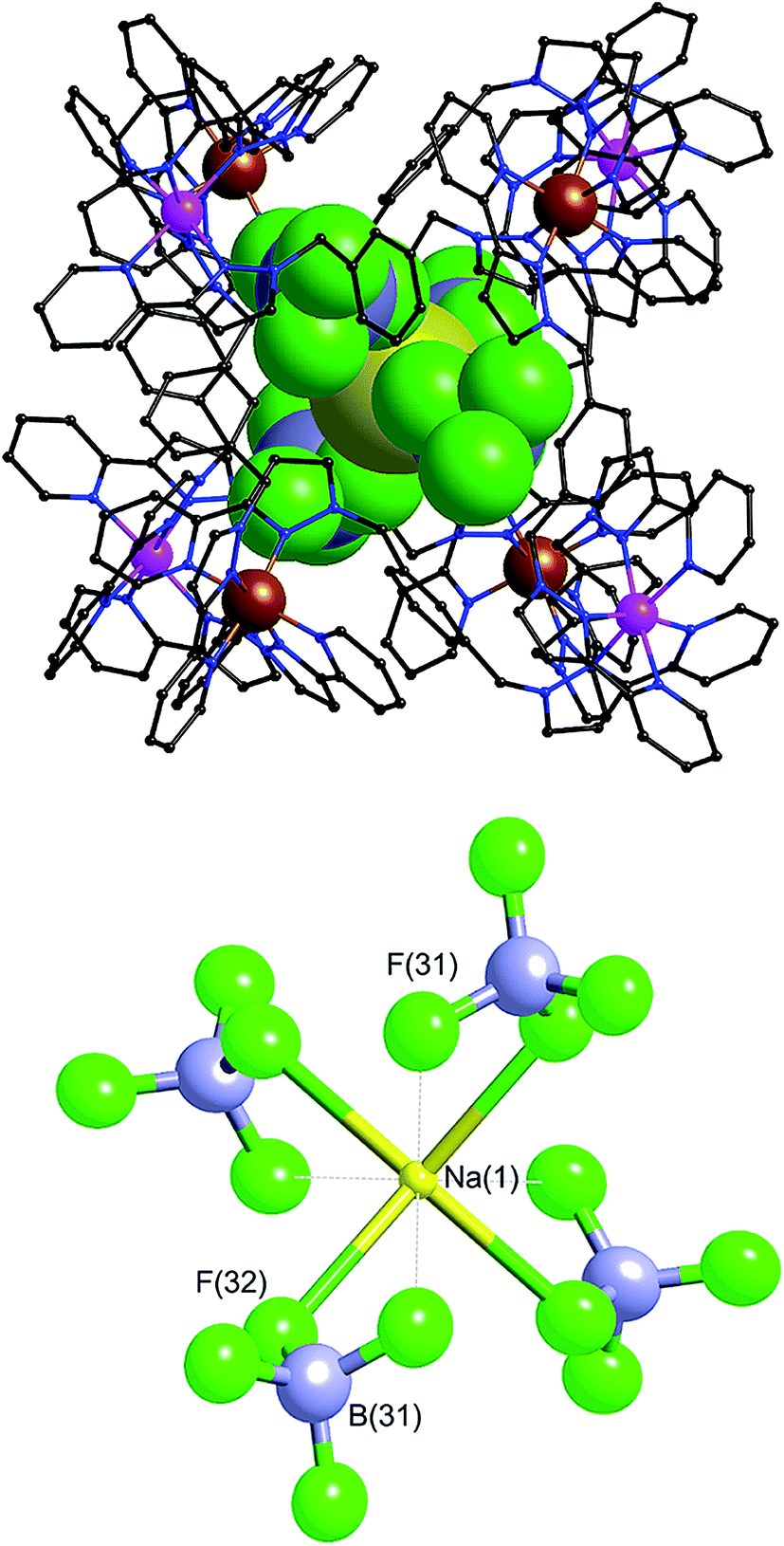 | ||
| Fig. 18 Two views of the structure of the complex [Ru4Co4(Lph)12{Na(BF4)4}](PF6)6(BF4)7. Left: View of the complete octanuclear cage (wireframe view), with the bound [Na(BF4)4]3− shown in space-filling mode. Right: Thermal ellipsoid plot of the bound guest [Na(BF4)4]3−, with ellipsoids shown at 40% probability. | ||
This octanuclear cage crystallised in the tetragonal space group P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 21m, with S4 molecular symmetry (axis through the centre of the face of the cube), such that one quarter of the complex cation is crystallographically unique. The asymmetric unit contains one Co(II) ion with a fac tris-chelate geometry and one Ru(II) ion with a mer tris-chelate geometry. This has the consequence of the complete cube having alternating fac (Co) and mer (Ru) metal sites around the framework, an arrangement which has not occurred in any previous cages of this family, even in the homonuclear analogues.8–10 Identification of the metal at each site turned out to be trivial; significantly different M–N bond lengths [average 2.07 Å (mer) and 2.13 Å (fac)] and physically unreasonable thermal parameters upon mislabelling confirmed that the mer site is occupied exclusively by Ru atoms, and the fac site by Co atoms, so the different metal types are now crystallographically ordered because of their different coordination geometry. Extensive π-stacking between the electron-rich and electron-deficient parts of adjacent ligands is clear around the periphery of the complex.
21m, with S4 molecular symmetry (axis through the centre of the face of the cube), such that one quarter of the complex cation is crystallographically unique. The asymmetric unit contains one Co(II) ion with a fac tris-chelate geometry and one Ru(II) ion with a mer tris-chelate geometry. This has the consequence of the complete cube having alternating fac (Co) and mer (Ru) metal sites around the framework, an arrangement which has not occurred in any previous cages of this family, even in the homonuclear analogues.8–10 Identification of the metal at each site turned out to be trivial; significantly different M–N bond lengths [average 2.07 Å (mer) and 2.13 Å (fac)] and physically unreasonable thermal parameters upon mislabelling confirmed that the mer site is occupied exclusively by Ru atoms, and the fac site by Co atoms, so the different metal types are now crystallographically ordered because of their different coordination geometry. Extensive π-stacking between the electron-rich and electron-deficient parts of adjacent ligands is clear around the periphery of the complex.
This new S4 structure for an M8L12 cubic cage is interesting in itself, but equally interesting is what lies inside the cavity. Usually with this family of cages, a solvent molecule or anion is found lying close to the convergent set of methylene protons surrounding the fac vertices, which form weak H-bond donor sites that can interact with electronegative atoms.9 As there are four fac tris-chelate vertices in this structure, there are potentially four recognition sites at which electron-rich guests may form hydrogen bonds with the interior surface of the cage. In this crystal structure, all of these sites are occupied.
Within the cavity there lie four tetrafluoroborate anions, one directed towards each fac vertex [around a Co(II) ion]. The organisation of these four anions into a tetrahedral array – dictated by the positioning of the four fac tris-chelate sites in the cube – results in formation of a central space surrounded by these four tetrafluoroborate anions – a ‘cavity within a cavity’, within which is bound a sodium cation which arises adventitiously (Fig. 18 and 19) and is most likely leached from the glassware. Two pieces of evidence support the assignment of the central atom as Na. Firstly, the distance to the nearest F atoms of the surrounding tetrafluoroborate anions is consistent with an Na⋯F dative interaction [Na(1)–F(32), 2.46 Å; Na(1)–F(31), 2.82 Å].13 Secondly, the thermal parameters become nonsensical when the atom is labelled differently (e.g. as K+ or Co2+). The arrangement of four anions in close proximity to one another inside the Ru4Co4 cage cavity is stabilised by coordination of all of them to Na+, as well as by numerous CH⋯F contacts between the ligands in the cage superstructure ligand and the encapsulated anions, the shortest of which is 2.23 Å between H(25C) and F(32).
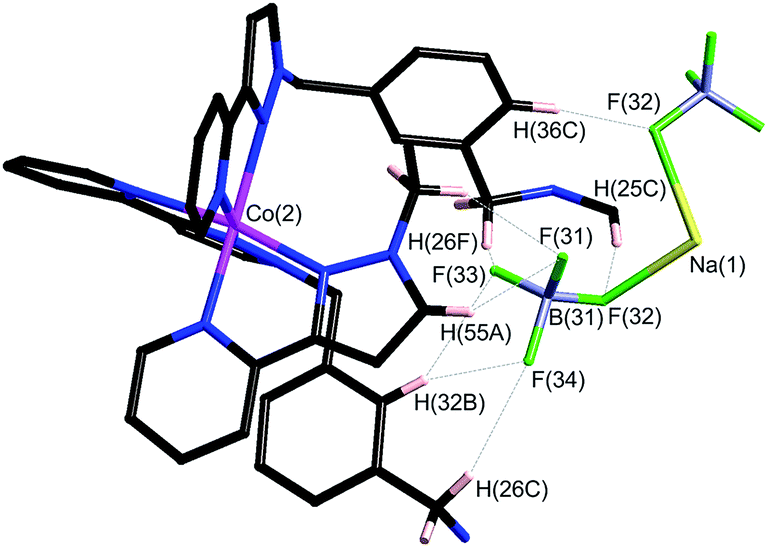 | ||
| Fig. 19 Partial view of one of the fac-[Co(Lph)3]2+ vertices in the [Ru4Co4(Lph)12]16+ complex cation, with CH⋯F interactions between ligands and the encapsulated (BF4)− anions indicated by dashed lines. | ||
Formation of this ‘complex within a complex’ requires three layers in a hierarchical self-assembly: the self-assembled Ru4Co4 cage encapsulates a tetrahedral array of four tetrafluoroborate anions, which in turn surround a central Na+ ion. This has parallels with the metallacrowns first reported by Pecoraro and co-workers,14 in which a transition-metal/ligand cyclic array based on Mn(III) ions and salicyl-hydroximate ligands results in an O-donor cavity whose structure is reminiscent of a crown ether, which accordingly coordinates additional alkali metal cations in the centre. It is also related to the observation from both Lindoy and co-workers15a and Nitschke and co-workers15b of the binding of tetrahalometallate anions as guests in the cavities of cationic M4L6 tetrahedral cage complexes. Addition of extra sodium salts to the crystallisation did not significantly improve the yield of crystalline material.
That this product should form containing exclusively the mer isomer of [Ru(Lph)3](PF6)2 can be rationalised on the basis that a large excess of this isomer was available for the cage-forming reaction. The minor product from the crystallisation (the orange shards) unfortunately did not yield any single crystals of sufficient quality to determine the crystal structure. The ES mass spectrum of these crystals revealed a mixture of tetranuclear [Ru2Co2(Lph)6]8+ and octanuclear [Ru4Co4(Lph)12]16+ species associated with varying numbers of anions. These may be presumed to incorporate the fac-[Ru(Lph)3](PF6)2 units in some form of assembly with Co(II) ions but could not be characterised further; the 1H NMR spectra were very complex and uninformative.
Finally we note that the difference in the nature of the products isolated by combination of [Ru(Lph)3](PF6)2 (4:1 mer:fac ratio) with Co(II) [which afforded the Ru3Co3 ‘open book’ as the only isolable crystalline product] and [Ru(Lph)3](PF6)2 (7:1 mer:fac ratio) with Co(II) [which afforded the new Ru4Co4 cube] cannot just be ascribed to the slightly higher proportion of the mer isomer of [Ru(Lph)3](PF6)2 in the latter case. The solvent systems used to grow the crystals were also different (MeCN/iPr2O in the former case; MeNO2/thf in the latter case) which could play an important role in determining which type of assembly is least soluble and therefore dominates the crystallisation.
Conclusions
We have explored two families of heteronuclear complexes in which pre-formed, kinetically stable [Ru(Lth)3]2+ and [Ru(Lph)3]2+ units (in the form of as-isolated mixtures of fac and mer isomers) are combined with labile M(II) ions (M = Co or Cd) to give Ru/M assemblies in which the Ru(II) and M(II) ions alternate in the metal array. With Lth as the bridging ligand we isolated the molecular square [Ru2Co2(Lth)6](BF4)4(PF6)4 and the one-dimensional coordination polymer {[CdRu(Lth)3](ClO4)2(PF6)2}∞. Both are based on heterodinuclear {RuM(Lth)2}4+ double helicate units, with two connected side by side by additional bridging ligand to form a Ru2Co2 molecular square; or a one-dimensional sequence linked end-to-end to give an alternating {RuCd}∞ chain.With Lph as the bridging ligand we isolated two quite different assemblies with Co(II) which contain different proportions of fac and mer Ru(II) units. These are the rectangular ‘open-book’ array [Ru3Co3(Lph)9](BF4)12 which contains a 2:1 proportion of fac/mer metal centres; and the cubic [Ru4Co4(Lph)12{Na(BF4)4}](PF6)6(BF4)7 cage which is a new structural type containing all mer Ru(II) vertices and all fac Co(II) vertices. The cavity of the cubic cage contains a tetrahedral array of fluoroborate anions which in turn are connected to a central Na(I) ion – a metal complex as the guest inside a metal complex.
Experimental section
General details
Metal salts and all organic reagents were purchased from Alfa or Sigma-Aldrich and used as received. NMR spectra were recorded on Bruker DRX 500 MHz, Bruker AV-III 400 MHz or AV-I 250 MHz instruments. Electrospray mass spectra were recorded on a Micromass LCT instrument. UV/Vis absorption spectra were measured on a Varian Cary 50 spectrophotometer. The ligands Lth and Lph were prepared according to the published methods.10,12 Ru(dmso)4Cl2 was prepared by the literature method.16Syntheses of mononuclear Ru(II) complexes
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 mer:fac ratio).
A solution of Lth (0.14 g, 0.35 mmol) was stirred rapidly in refluxing ethylene glycol (50 cm3) until dissolved. To this was added a solution of RuCl2(dmso)4 (0.02 g, 0.04 mmol) in H2O/ethylene glycol (3:100, 103 cm3) by dropping funnel over 3 hours, and then the orange mixture was stirred at reflux for 14 h. The solution was cooled to 25 °C and excess saturated KPF6(aq.) was added. The product was extracted with dichloromethane, dried over MgSO4, and evaporated to dryness.
:1 mer:fac ratio).
A solution of Lth (0.14 g, 0.35 mmol) was stirred rapidly in refluxing ethylene glycol (50 cm3) until dissolved. To this was added a solution of RuCl2(dmso)4 (0.02 g, 0.04 mmol) in H2O/ethylene glycol (3:100, 103 cm3) by dropping funnel over 3 hours, and then the orange mixture was stirred at reflux for 14 h. The solution was cooled to 25 °C and excess saturated KPF6(aq.) was added. The product was extracted with dichloromethane, dried over MgSO4, and evaporated to dryness.
The product was purified by column chromatography on silica. Elution with MeCN–water–saturated aqueous KNO3 (100:10:1, v/v) resulted in two yellow bands moving down the column – the second, major band was collected. After removing acetonitrile by rotary evaporation, excess saturated aqueous KPF6 was added and the product was extracted from the resulting suspension into dichloromethane. The organic layer was separated, dried over MgSO4, and the solvent removed in vacuo to yield [Ru(Lth)3](PF6)2, 3:1 mer:fac ratio, as a yellow solid. Yield: 0.04 g, 54%.
ESMS: m/z 648 (M − 2PF6)2+, 432 (M − 2PF6 + H)3+. UV/Vis in MeCN [λmax/nm (10−3ε/M−1 cm−1)]: 396 (13.1), 281 (72.8), 243 (81.5). Found: C, 48.2; H, 3.9; N, 15.1%. Required for C66H54N18P2F12RuS3·3H2O: C, 48.3; H, 3.7; N, 15.4%.
:1 mer:fac ratio).
A solution of Lph (0.30 g, 0.76 mmol) in ethanol (100 cm3) and H2O (20 cm3) was stirred under reflux until fully dissolved. To this was added a solution of RuCl2(dmso)4 (0.05 g, 0.11 mmol) in ethanol/H2O (7:5 v/v, 60 cm3) by dropping funnel over 3 hours, and then the yellow mixture was stirred at reflux in the dark for 14 h. After cooling the red mixture and diluting with H2O, excess ligand was removed by washing with chloroform. Addition of saturated KPF6(aq.) afforded a yellow precipitate, which was purified by column chromatography on silica. Elution with MeCN–water–saturated aqueous KNO3 (100:10:1 v/v) resulted in a broad yellow band moving down the column which was collected. After removing acetonitrile by rotary evaporation, excess saturated aqueous KPF6 was added and the product was extracted from the suspension into dichloromethane. The organic layer was separated, dried over MgSO4, and the solvent removed in vacuo to yield [Ru(Lph)3](PF6)2 as a yellow solid (7:1 mer:fac ratio). Yield: 0.24 g, 78%. ESMS m/z 1423 (M − PF6)+, 639 (M − 2PF6)2+, 426 (M + H − 2PF6)3+. Found: C, 54.7; H, 4.1; N, 15.8%. Required for C72H60N18P2F12Ru: C, 55.1; H, 3.9; N, 16.1%.
Synthesis of polynuclear heterometallic complexes
:1 mixture of mer:fac isomers; 0.073 g, 0.047 mmol) in dichloromethane (10 cm3) was added a solution of Co(BF4)2·6H2O (0.016 g, 0.047 mmol) in methanol (10 cm3). After an overnight stir, the mixture was evaporated to dryness and then washed with dichloromethane and methanol. The mixture was then collected on a membrane filter and then extracted with nitromethane. Slow diffusion of tetrahydrofuran vapour into the nitromethane solution over two months gave the product as yellow blocks in 35% isolated yield, and orange shards in 14% isolated yield. ESMS (selected peaks): m/z 1757, ([Ru4Co4(Lph)12](BF4)(PF6)11)4+; 1743, ([Ru4Co4(Lph)12](BF4)2(PF6)10)4+; 1377, ([Ru4Co4(Lph)12](BF4)(PF6)10)5+; 1365, ([Ru4Co4(Lph)12](BF4)2(PF6)9)5+; 1123, ([Ru4Co4(Lph)12](BF4)(PF6)9)6+; 1114, ([Ru4Co4(Lph)12](BF4)2(PF6)8)6+; 942, ([Ru4Co4(Lph)12](BF4)(PF6)8)7+; 934, ([Ru4Co4(Lph)12](BF4)2(PF6)7)7+; 806, ([Ru4Co4(Lph)12](BF4)(PF6)7)8+; 799, ([Ru4Co4(Lph)12](BF4)2(PF6)6)8+. Elemental analytical data was consistent with the presence of water of crystallisation due to the desolvated material being hygroscopic. Found: C, 46.2; H, 3.6; N, 13.0%. Required for C288H240B9NaCo4F84N72P8Ru4·12H2O: C, 45.9; H, 3.5; N, 13.4%.
:1 mixture of mer:fac isomers; 0.053 g, 0.034 mmol) in dichloromethane (10 cm3) was added a solution of Co(BF4)2·6H2O (0.126 g, 0.370 mmol) in methanol (10 cm3). After an overnight stir, the mixture was evaporated to dryness and then washed with dichloromethane and methanol on a membrane filter. Slow diffusion of diisopropyl ether into a solution of the solid in nitromethane gave the product as yellow laths in low yield (17%). ESMS (selected peaks): m/z 2440, ([Ru3Co3(Lph)9](BF4)10)2+; 1599, ([RuCo(Lph)3](BF4)3)+; 1177, ([Ru3Co3(Lph)9](BF4)8)4+; 1036, ([Ru2Co2(Lph)6](BF4)5)3+; 924, ([Ru3Co3(Lph)9](BF4)7)5+; 756, ([RuCo(Lph)3](BF4)2)2+; 452, ([RuCo(Lph)3]F)3+. UV/Vis in MeCN [λmax/nm (10−3ε/M−1 cm−1)]: 396 (38.7), 282 (216.1), 244 (206.7).
X-ray crystallography
Diffraction data for the structures [Ru4Co4(Lph)12]Na(BF4)11(PF6)6·8MeNO2 and 2{[Ru3Co3(Lph)9](BF4)12}·11MeNO2·3H2O were collected on a Bruker Apex-II diffractometer at the University of Sheffield. Diffraction data for 2{[Ru2Co2(Lth)6]}(BF4)10(PF6)6·5MeCN·2H2O were collected by the National Crystallographic Service using a synchrotron radiation source.17 In each case a crystal was removed from the mother liquor, coated with oil, and transferred rapidly to a stream of cold N2 on the diffractometer to prevent rapid decomposition due to solvent loss which occurred in all cases. In all cases, after integration of the raw data, and before merging, an empirical absorption correction was applied (SADABS)18 based on comparison of multiple symmetry-equivalent measurements. The structures were solved by direct methods and refined by full-matrix least squares on weighted F2 values for all reflections using the SHELX suite of programs.19 Pertinent crystallographic data are collected in Table 1.| Complex | 2{[Ru2Co2(Lth)6]}(BF4)10(PF6)6·5MeCN·2H2O | [Ru4Co4(Lph)12]Na(BF4)11(PF6)6·8MeNO2 | 2{[Ru3Co3(Lph)9](BF4)12}·11MeNO2·3H2O |
|---|---|---|---|
| a The compositions are necessarily approximate as they do not include solvent molecules eliminated from the refinement as part of the ‘SQUEEZE’ process. b The value of R1 is based on ‘observed’ data with I > 2σ(I); the value of wR2 is based on all data. | |||
| Formula | C274H235B10Co4F76 N77P6Ru4S12 | C296H264B11Co4F80N80NaO16P6Ru4 | C443H399B24Co6F96N119 O25Ru6 |
| Molecular weight | 7401.02 | 7685.58 | 10833.23 |
| T, K | 100(2) | 100(2) | 100(2) |
| Crystal system | Monoclinic | Tetragonal | Triclinic |
| Space group | P21/c |
P21m |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a, Å | 17.3091(12) | 31.3553(12) | 26.036(5) |
| b, Å | 42.874(3) | 31.3553(12) | 27.068(6) |
| c, Å | 22.2336(16) | 21.7210(8) | 42.869(10) |
| α, ° | 90 | 90 | 84.340(5) |
| β, ° | 103.1010(18) | 90 | 88.764(5) |
| γ, ° | 90 | 90 | 87.336(8) |
| V, Å3 | 16070(2) |
21355.1(18) |
30027(11) |
| Z | 2 | 2 | 2 |
| ρ, g cm−3 | 1.529 | 1.195 | 1.198 |
| Crystal size, mm3 | 0.07 × 0.03 × 0.01 | 0.2 × 0.2 × 0.2 | 0.32 × 0.16 × 0.08 |
| μ, mm−1 | 0.556 | 0.403 | 0.396 |
| Independent data, restraints, parameters | 17217/139/1901 |
15202/832/840 |
62175/302/1895 |
| Final R1, wR2b | 0.1088, 0.2800 | 0.0794, 0.2209 | 0.1289, 0.3298 |
In all cases crystals exhibited the usual problems of this type of structure, viz. weak scattering due to a combination of poor crystallinity, solvation, and disorder of anions/solvent molecules. In each case the basic structure and connectivity of the complex cation could be unambiguously determined with reasonable precision. Extensive use of geometric restraints on aromatic rings and anions, and restraints on aromatic displacement parameters, were required to keep refinements stable. Solvent molecules and anions that could be modelled satisfactorily were included in the final refinements; in all cases large regions of diffuse electron density that could not be modelled (from disordered solvents/counter ions) were removed from the refinement, using the SQUEEZE function in PLATON.20 Full details of these issues and how they were handled are given in the individual CIFs; it should be noted that the compositions/formulae of the crystals as given in Table 1 are necessarily an approximation. CCDC deposition numbers: 1433701–1433703.†
Acknowledgements
We thank the EPSRC for financial support (grant EP/K003224/1), and Dr Andrew Stephenson for synthesis of a sample of Lth.References
- (a) D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 349 CrossRef CAS PubMed; (b) M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369 CrossRef CAS PubMed; (c) M. D. Ward, Chem. Commun., 2009, 4487 RSC; (d) J. J. Perry, J. A. Perman and M. J. Zaworotko, Chem. Soc. Rev., 2009, 38, 1400 RSC; (e) H. Amouri, C. Desmarets and J. Moussa, Chem. Rev., 2012, 112, 2015 CrossRef CAS PubMed; (f) A. F. Williams, Coord. Chem. Rev., 2011, 255, 2104 CrossRef CAS; (g) Z. Laughrey and B. Gibb, Chem. Soc. Rev., 2011, 40, 363 RSC; (h) P. Jin, S. J. Dalgarno and J. L. Atwood, Coord. Chem. Rev., 2012, 254, 1760 CrossRef; (i) R. J. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 CrossRef CAS PubMed; (j) Y. Inokuma, M. Kawano and M. Fujita, Nat. Chem., 2011, 3, 349 CrossRef CAS PubMed; (k) M. D. Pluth, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2009, 42, 1650 CrossRef CAS PubMed; (l) M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728 RSC; (m) T. Nakamura, H. Ube and M. Shionoya, Chem. Lett., 2014, 42, 328 CrossRef; (n) H. Li, Z.-J. Yao, D. Liu and G.-X. Jin, Coord. Chem. Rev., 2015, 293–294, 139 CrossRef CAS; (o) L. Chen, Q. Chen, M. Wu, F. Jiang and M. Hong, Acc. Chem. Res., 2015, 48, 201 CrossRef CAS PubMed; (p) T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001 CrossRef CAS PubMed; (q) L. Li, D. J. Fanna, N. D. Shepherd, L. F. Lindoy and F. Li, J. Inclusion Phenom. Macrocyclic Chem., 2015, 82, 3 CrossRef CAS.
- M. D. Ward and P. R. Raithby, Chem. Soc. Rev., 2013, 42, 1619 RSC.
- (a) J. W. Yi, N. P. E. Barry, M. A. Furrer, O. Zava, P. J. Dyson, B. Therrien and B. H. Kim, Bioconjugate Chem., 2012, 23, 461 CrossRef CAS PubMed; (b) B. Therrien, Chem.–Eur. J., 2013, 19, 8378 CrossRef CAS PubMed; (c) J. E. M. Lewis, E. L. Gavey, S. A. Cameron and J. D. Crowley, Chem. Sci., 2012, 3, 778 RSC; (d) W. Cullen, S. Turega, C. A. Hunter and M. D. Ward, Chem. Sci., 2015, 6, 625 RSC.
- (a) C. J. Brown, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2009, 131, 17530 CrossRef CAS PubMed; (b) C. J. Hastings, M. D. Pluth, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2010, 132, 6938 CrossRef CAS PubMed; (c) J. L. Bolliger, A. M. Belenguer and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 7958 CrossRef CAS PubMed; (d) H. Vardhan and F. Verpoort, Adv. Synth. Catal., 2015, 357, 1351 CrossRef CAS; (e) X. Jing, C. He, Y. Yang and C. Duan, J. Am. Chem. Soc., 2015, 137, 3967 CrossRef CAS PubMed; (f) C. García-Simón, R. Gramage-Doria, S. Raoufmoghaddam, T. Parella, M. Costas, X. Ribas and J. N. H. Reek, J. Am. Chem. Soc., 2015, 137, 2680 CrossRef PubMed; (g) C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012 CrossRef CAS PubMed.
- (a) O. Chepelin, J. Ujma, X. Wu, A. M. Z. Slawin, M. B. Pitak, S. J. Coles, J. Michel, A. C. Jones, P. E. Barran and P. J. Lusby, J. Am. Chem. Soc., 2012, 134, 19334 CrossRef CAS PubMed; (b) K. Yamashita, M. Kawano and M. Fujita, Chem. Commun., 2007, 4102 RSC; (c) Z. Li, N. Kishi, K. Hasegawa, M. Akita and M. Yoshizawa, Chem. Commun., 2011, 47, 8605 RSC; (d) X. Yan, T. R. Cook, P. Wang, F. Huang and P. J. Stang, Nat. Chem., 2015, 7, 342 CrossRef CAS PubMed; (e) L.-L. Yan, C.-H. Tan, G.-L. Zhang, L.-P. Zhou, J.-C. Bünzli and Q.-F. Sun, J. Am. Chem. Soc., 2015, 137, 8550 CrossRef CAS PubMed; (f) C. He, Z. Lin, C. Duan, C. Xu, Z. Wang and C. Yan, Angew. Chem., Int. Ed., 2008, 47, 877 CrossRef CAS PubMed; (g) P. D. Frischmann, V. Kinz and F. Würthner, Angew. Chem., Int. Ed., 2015, 54, 7285 CrossRef CAS PubMed; (h) P. P. Neelakandan, A. Jiménez and J. R. Nitschke, Chem. Sci., 2015, 5, 908 RSC.
- (a) A. J. Metherell and M. D. Ward, Chem. Commun., 2014, 50, 6330 RSC; (b) A. B. Wragg, A. J. Metherell, W. Cullen and M. D. Ward, Dalton Trans., 2015, 44, 17869 RSC; (c) A. J. Metherell and M. D. Ward, Chem. Commun., 2014, 50, 10979 RSC; (d) A. J. Metherell and M. D. Ward, Polyhedron, 2015, 89, 260 CrossRef CAS.
- (a) Y.-T. Chan, X. Li, J. Yu, G. A. Carri, C. N. Moorefield, G. R. Newkome and C. Wesdemiotis, J. Am. Chem. Soc., 2011, 133, 11967 CrossRef CAS PubMed; (b) H. Sato, A. Nakao and A. Yamagishi, New J. Chem., 2011, 35, 1823 RSC; (c) M. M. J. Smulders, A. Jimenez and J. R. Nitschke, Angew. Chem., Int. Ed., 2012, 51, 6681 CrossRef CAS PubMed; (d) K. Li, L.-Y. Zhang, C. Yan, M. Pan, L. Zhang and C.-Y. Su, J. Am. Chem. Soc., 2014, 136, 4456 CrossRef CAS PubMed; (e) A. Galstyan, P. J. Sanz Miguel, K. Weise and B. Lippert, Dalton Trans., 2013, 42, 16151 RSC; (f) P. de Wolf, S. L. Heath and J. A. Thomas, Chem. Commun., 2002, 2540 RSC; (g) D. M. Bassani, J.-M. Lehn, S. Serroni, F. Puntoriero and S. Campagna, Chem.–Eur. J., 2003, 9, 5936 CrossRef CAS PubMed; (h) J. Yang, M. Bhadbhade, W. A. Donald, H. Iranmanesh, E. G. Moore, H. Yan and J. E. Beves, Chem. Commun., 2015, 51, 4465 RSC; (i) A. J. Metherell and M. D. Ward, Chem. Sci. 10.1039/c5sc03526k.
- (a) X. Sun, D. W. Johnson, D. L. Caulder, K. N. Raymond and E. H. Wong, J. Am. Chem. Soc., 2001, 123, 2752 CrossRef CAS PubMed; (b) S. Hiraoka, Y. Sakata and M. Shionoya, J. Am. Chem. Soc., 2008, 130, 10058 CrossRef CAS PubMed; (c) W. J. Ramsay, T. K. Ronson, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 13439 CrossRef CAS PubMed; (d) F. E. Hahn, M. Offermann, C. SchulzeIsfort, T. Pape and R. Frohlich, Angew. Chem., Int. Ed., 2008, 47, 6794 CrossRef CAS PubMed; (e) H.-B. Wu and Q.-M. Wang, Angew. Chem., Int. Ed., 2009, 48, 7343 CrossRef CAS PubMed; (f) A. J. Metherell and M. D. Ward, RSC Adv., 2013, 3, 14281 RSC.
- (a) I. S. Tidmarsh, T. B. Faust, H. Adams, L. P. Harding, L. Russo, W. Clegg and M. D. Ward, J. Am. Chem. Soc., 2008, 130, 15167 CrossRef CAS PubMed; (b) S. Turega, M. Whitehead, B. R. Hall, M. F. Haddow, C. A. Hunter and M. D. Ward, Chem. Commun., 2012, 48, 2752 RSC; (c) S. Turega, M. Whitehead, B. R. Hall, A. J. H. M. Meijer, C. A. Hunter and M. D. Ward, Inorg. Chem., 2013, 52, 1122 CrossRef CAS PubMed; (d) M. Whitehead, S. Turega, A. Stephenson, C. A. Hunter and M. D. Ward, Chem. Sci., 2013, 4, 2744 RSC; (e) S. Turega, W. Cullen, M. Whitehead, C. A. Hunter and M. D. Ward, J. Am. Chem. Soc., 2014, 136, 8475 CrossRef CAS PubMed.
- A. Stephenson and M. D. Ward, Dalton Trans., 2011, 40, 10360 RSC.
- S. L. Dabb and N. C. Fletcher, Dalton Trans., 2015, 44, 4406 RSC.
- (a) S. P. Argent, H. Adams, L. P. Harding and M. D. Ward, Dalton Trans., 2006, 542 RSC; (b) A. M. Najar, I. S. Tidmarsh, H. Adams and M. D. Ward, Inorg. Chem., 2009, 48, 11871 CrossRef CAS PubMed.
- B. Whittle, S. R. Batten, J. C. Jeffery, L. H. Rees and M. D. Ward, J. Chem. Soc., Dalton Trans., 1996, 4249 RSC.
- (a) M. S. Lah and V. L. Pecoraro, J. Am. Chem. Soc., 1989, 111, 7258 CrossRef CAS; (b) A. J. Stemmler, J. W. Kampf and V. L. Pecoraro, Inorg. Chem., 1995, 34, 2271 CrossRef CAS; (c) A. J. Stemmler, A. Barwinski, M. J. Baldwin, V. Young and V. L. Pecoraro, J. Am. Chem. Soc., 1996, 118, 11962 CrossRef CAS.
- (a) C. R. K. Glasson, J. K. Clegg, J. C. McMurtrie, G. V. Meehan, L. F. Lindoy, C. A. Motti, B. Moubaraki, K. S. Murray and J. D. Cashion, Chem. Sci., 2011, 2, 540 RSC; (b) I. A. Riddell, T. K. Ronson and J. R. Nitschke, Chem. Sci., 2015, 6, 3533 RSC.
- I. P. Evans, A. Spencer and G. Wilkinson, J. Chem. Soc., Dalton Trans., 1973, 204 RSC.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683 RSC.
- G. M. Sheldrick, SADABS: A program for absorption correction with the Siemens SMART system, University of Göttingen, Germany, 2008 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- (a) A. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS; (b) P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1433701–1433703. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra22694e |
| This journal is © The Royal Society of Chemistry 2016 |